 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ernesto Cuenca Zamora | + 2408 word(s) | 2408 | 2021-01-28 04:11:54 | | | |
| 2 | Dean Liu | + 1 word(s) | 2409 | 2021-02-19 07:25:51 | | |
Video Upload Options
Neutrophils, the major innate immune cells, eliminate pathogens by phagocytosis or by releasing antimicrobial proteolytic enzymes present in their granules.
1. Introduction
Neutrophils, the major innate immune cells, eliminate pathogens by phagocytosis or by releasing antimicrobial proteolytic enzymes present in their granules. In recent years, another strategy by which neutrophils kill pathogens has been identified and named NETosis[1]. NETs are extracellular structures composed of DNA and histones (nucleosomes) associated with antibacterial proteins (including myeloperoxidase, elastase, pentraxin, matrix metalloproteinase 9 (MMP9)) that entrap, immobilize and kill pathogens aiding against infections[1][2].
NETs formation is a dynamic process (Figure 1). Upon activation, neutrophils adhere to the endothelium and granular enzymes (myeloperoxidase and elastase) are translocated into the nucleus. The latter, together with the activation of the enzyme peptidyl arginine deaminase 4 (PAD4) promote the de-condensation of chromatin, the loss of the lobular form of the neutrophil[3][4], and the rupture of its nuclear membrane. Granular proteins bound to chromatin are expelled into the extracellular space with or without rupture of the plasma membrane -processes called suicidal or vital NETosis, respectively. In vital NETosis, neutrophils survive NET release and can continue to phagocytize pathogens [5][6]. The suicidal NETosis, by contrast, is considered a specific form of cellular death, dependent on the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and the generation of ROS[7]. Whether one mechanism or the other is induced depends on the stimulus that triggers the process[5][6][8]: pathogens (bacteria, fungi, viruses, and protozoa) in the vital NETosis or inflammatory stimuli (LPS, IL-8, TNFα), activated platelets, auto-antibodies, or cholesterol crystals in the suicidal (or sterile) NETosis. In both vital and suicidal NETosis, PAD4-mediated histone citrullination is thought to promote NETs formation by inducing chromatin decondensation, facilitating the expulsion of chromosomal DNA[3][4]. Thus, PAD4 is essential in the NETs formation, and PAD4-deficient mice are unable to generate NETs[3][4].
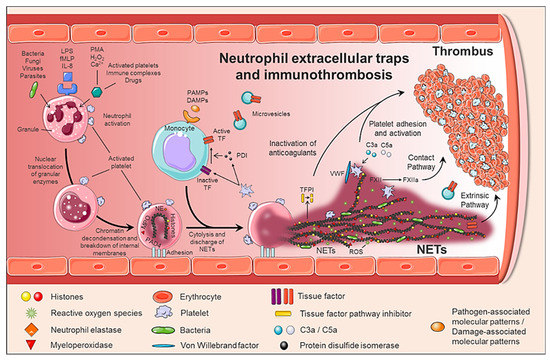
Figure 1. Generation of neutrophil extracellular traps (NETs) and immunothrombosis concept. The formation of NETs is a dynamic and complex process, in which not only neutrophils are involved, but also other circulating cells such as monocytes and platelets. When activated, neutrophils adhere to the endothelium and granular enzymes (myeloperoxidase and elastase) are translocated to the nucleus, which together with the activation of PAD4 promotes the decongestion of chromatin, the loss of the lobular form of the neutrophil, and the rupture of its nuclear membrane. Finally, granular proteins bound to chromatin are expelled into the extracellular space (NETs), providing a perfect structure not only to immobilize and kill pathogens, but also to induce a pro-coagulant response.
Although NETs play a critical role in immune defense, excessive formation, or ineffective elimination can result in unwanted adverse effects. Therefore, its degradation is an important physiological process carried out by DNase I. Although the mechanisms involved in the clearance of NETs are not yet fully understood, macrophages also participate in the clearance of NETs by endocytic processes[2][9].
2. Role of NETs in Thrombotic Pathogenesis
NETs therefore not only serve as mediators of neutrophil antibacterial functions but also provide a scaffold for inducing a strong procoagulant response. Engelmann and Massberg introduce the term “immunothrombosis” to describe the link between innate immunity and thrombosis (Figure 2). It is based on the capacity of the NET to induce a procoagulant response that leads to the formation of a thrombus as a physiological defense mechanism against pathogens[10]. “Immunothrombosis is supported by immune cells and by thrombosis-related molecules”[10][11]:
-
Any cell death is a potential source of free DNA in plasma, so this is a necessary but not specific finding of NETosis. Although not specific to NETosis, the presence of negative charges (DNA) causes an activation of FXII, a plasma serine protease, initiating the intrinsic pathway of coagulation. This promotes the chain activation of a series of coagulation proteins which in turn results in the formation of fibrin and ultimately the thrombus[12];
-
Histones are the most abundant proteins in NETs. They are positively charged and are responsible for packaging the genetic material. It has been shown that histones 3 and 4 (H3 and H4, respectively) are able to activate platelets, favoring their aggregation and contributing to the generation of thrombin[13]. This ability of H3 and H4 to activate platelets seems to be, at least partially, dependent on the signaling pathway of TLR2 and TLR4 receptors, through the transcription factor NF-κB[13]. Alternatively, histones also contribute to thrombin activation by reducing thrombomodulin-dependent protein C activation[14];
-
The granule proteases (elastase and Cathepsin G) are enzymes derived from neutrophils and the most abundant proteins in NET after histones. Elastase is located in the acidophilic granules and its function is to eliminate tissue degradation products of pathogens. In the context of thrombogenesis, it causes the degradation and inactivation of two important natural anticoagulants: tissue factor pathway inhibitor (TFPI) and antithrombin (AT)[15][16]. TFPI is the main inhibitor of the TF pathway or extrinsic pathway of coagulation whereas AT blocks thrombin formation, a key step for thrombus formation. In addition, elastase promotes platelet adhesion by facilitating exposure to von Willebrand factor (vWF). Cathepsin G hydrolyzes proteins and also helps block the activity of TFPI and enhance thrombosis by activating the protease receptor 4 (PAR4) signaling pathway on platelets. Thus, it was observed that mice deficient in elastase and cathepsin G have defects in TF activation, in fibrin formation, and in thrombus stabilization [17];
-
TF, through activation of the extrinsic pathway of coagulation and platelets, promotes thrombus formation. TF has been identified in NETs and it has been documented that this factor comes not only from monocytes that migrate to the inflamed area, but also from neutrophils. This finding was observed in neutrophils isolated from patients with sepsis. Autophagy has been pointed out as the mechanism by which the neutrophil captures the TF that is released during NETosis. In this sense, the TF carried by NETs is capable of stimulating thrombin generation and platelet activation in ex vivo experiments[18][19].
Altogether, NETs generate an intravascular scaffold, according to which the fibrin network facilitates the recognition, containment and destruction of pathogens[10]; the microthrombus prevents the invasion of pathogens through the circulation and generates a compartment where antimicrobial substances are concentrated for greater effectiveness; finally, the accumulation of fibrinogen or fibrin deposits promotes the recruitment of other immune cells by coordinating the immune response[10][11].
3. NETs in Vascular Pathology
Recently, the number of pathologies in which NETs can play a relevant role is increasing. There is an increasing amount of clinical and experimental data supporting the role of NETs in a wide variety of pathological conditions, both infectious and non-infectious[5]. Thus, the presence of NETs in autoimmune diseases, diabetes, atherosclerosis, vasculitis, and cancer has been pointed out. Furthermore, uncontrolled production of NETs in blood vessels may constitute a decisive biological basis for the development of thrombotic events, including venous thrombosis, arterial thrombosis, and microvascular thrombosis[11][17][20].
Different physiopathological processes normally trigger thrombotic events, both venous and arterial, although they share common risk factors. Thus, in arterial thrombosis, the activation, aggregation, and adhesion of platelets to the endothelial wall play a very important role, ultimately leading to the formation of so-called “white” platelet-rich thrombi. In contrast, a key factor for venous thrombosis is a reduction in blood flow and activation of circulating coagulation factors, which results in ”red” thrombi due to the local accumulation of large numbers of red blood cells.
The participation of NETs in arterial, venous and microvascular thrombosis has been validated both in animal models and in clinical studies[21][22][23]. Inferior vena cava and iliac vein stenosis in mice and baboons, respectively, demonstrated the presence of NETs associated with vWF within the venous thrombus and an increase of NETs markers in plasma[22][24]. In addition, the injection of extracellular histones promotes the development of DVT, while the administration of DNase I attenuates it [24]. NETs have also been identified in human venous thrombi and in plasma of patients with DVT and venous thromboembolism, being associated with increased thrombotic risk[25]. Indeed, sera and plasma from patients with primary antiphospholipid syndrome (PAPS), who carry a markedly increased risk of thrombotic events and pregnancy loss, showed elevated levels of NETs, as compared to healthy volunteers[26]. Specifically, administration of IgG from these patients accelerates venous thrombosis in a flow restriction murine model, a phenotype that associates with human IgG binding to the neutrophil surface and with an expanded infiltration of NETs into the thrombi themselves[27].
Moreover, recent studies have shown how NETs contribute to the initiation and progression of atherosclerotic lesions and arterial thrombus growth[28]. Thus, in a murine model of atherosclerosis, PAD4 inhibition was able to prevent the formation of NETs, decrease the size of the atherosclerotic lesion, and delay carotid artery thrombosis[29]. Another work performed on ApoE−/− mice showed that cholesterol crystals (sterile stimulus) have the capacity to generate NETs that activated macrophages, amplifying cell recruitment in the lesion area[30]. In humans, the presence of NETs has been associated with coronary atherosclerosis and myocardial infarction[31].
Finally, patients with sepsis and/or dissemin[32]ated intravascular coagulation have elevated TF levels in monocytes, leukocyte-platelet aggregates, and increased levels of NETs markers[33][34].
4. Role of NETs in Myeloproliferative Neoplasms
As previously mentioned, NETs appear on both infectious and non-infectious diseases, e.g., autoimmune disease or cancer, under the stimulus of cytokines (TNFα and IL-8) secreted by the neoplasm clone itself[35] or by activated platelets[36][37][38]. When platelets are activated, P-selectin is translocated to the membrane from α-granules. P-selectin, both cellular and soluble, promotes NETosis through binding to PSGL-1. The process can be inhibited by blocking either P-selectin or PSGL-1. Indeed, activated platelets from P-selectin null mice were unable to trigger NETs, whereas neutrophils from mice engineered to overproduce soluble P-selectin had excessive agonist-induced NETs formation, suggesting that the P-selectin/PSGL-1 axis is a potential therapeutic target [73].
Although it is a physiological process, uncontrolled production of NETs may constitute the basis for the development of thrombotic disorders[11][17]. In a recent prospective observational cohort study with nearly 1000 cancer patients and two years of follow-up, citrullinated H3 (citH3), a biomarker of NET formation, predicted the risk of venous thromboembolism. Thus, citH3 levels had a magnitude of association with venous thromboembolism risk comparable to D-Dimer or soluble P-selectin[38].
Specifically, three studies have evaluated whether NETs contribute to the procoagulant state in MPN patients [39][40][41]. Although it seems obvious that the percentage of neutrophils with increased levels of ROS is higher in patients with MPN than controls[40][41], it is not clear if under baseline conditions, i.e., without stimulation, they produce more NETs. Whereas Guy et al. showed that unstimulated neutrophils from patients with MPN, ex vivo, produced more NETs than control subjects[41], Oyarzún et al. and Wolach et al., in two independent studies did not find enhanced NETosis by unstimulated JAK2V617F neutrophils[39][40]. These contradictory results have been attributed to the fact that in the last two cohorts of patients[39][40], most of the patients were receiving JAK inhibitors or cytoreductive treatment at the time of inclusion in the study. Other potential biases that may explain these contradictory results could be derived from the small number of patients included for this purpose in 2 out of the 3 studies (n = 19 and 32 patients in Oyarzún et al. and Wolach et al. studies, respectively), and the enrichment of patients with previous thrombosis in the third of these studies (26 out of 52, 50% of patients in Guy’s cohort)[41]. One additional explanation is the non-standardized assays used to assess NETosis[39][40][41]. In fact, there are still no methods to assess and determine NETosis in a reproducible and objective manner[42].
Regarding the formation of NETs ex vivo, under stimulation, the results are also contradictory. Ex vivo stimulation of neutrophils with ionomycin, caused an increase in NET formation (citH3 expression) in both JAK2V617F human and mouse neutrophils[39]. By contrast, another study did not find NETs production after stimulation with IL-8, or TNFα; and with a stronger NETs inducer, such as PMA, MPN cells showed defective NETosis[40].
Moreover, in two independent cohorts of patients with MPN, the evaluation of plasmatic biomarkers of NETosis has shown an increase in the concentration of free plasma DNA[41] and elevated levels of circulating nucleosomes[40], another DNA marker. However, free plasma DNA or nucleosomes are not specific markers of NETs, they can be originated also from other forms of cell death, such as apoptosis or necrosis. More specific markers of NETs combine measurement of DNA (nucleosomes, histones, or free DNA) with a specific enzyme (myeloperoxidase o elastase) from neutrophils. While Oyarzún et al. did not find higher levels of histone-MPO in MPN patients as compared to healthy donors[40], Guy et al. showed a significant increase in MPO-DNA concentration in patients with MPN at the time of presentation compared to controls[41]. Importantly, MPO-DNA levels were higher in MPN patients with previous thrombosis, especially with splenic thrombosis, positioning itself as a biomarker of thrombosis in patients with MPN[41].
In regards to the effect of cytoreductors on NET markers, ruxolitinib abrogates NETs formation ex vivo (in neutrophils from patients receiving the JAK1/2 inhibitor) and, in vivo, decreasing thrombosis in JAK2V617F mice[39]. Oyarzún et al. showed that both hydroxyurea and ruxolitinib decrease the concentration of nucleosomes[40]. By contrast, Guy et al. (in 10 patients) did not find that treatments were associated with a decrease of free DNA or MPO-DNA complexes, despite the normalization of neutrophils counts[41].
In JAK2V617F/WT; Vav-Cre mice, with heterozygous expression of the JAK2V617F allele in hematopoietic cells (JAK2V617F), Wolach et al. demonstrated an increased lung thrombi formation. To further explore the role of NETosis in MPN thrombosis, these authors investigated the development of thrombosis in an experimental model of NET-dependent thrombosis in JAK2V617F mice[39]. Two hours after partial ligation of the inferior vena cava, 45% of the JAK2V617F mice developed thrombosis while none of the JAK2 wild type (JAK2WT) mice. The treatment during 72 h with ruxolitinib reduced the range of thrombosis to levels comparable to JAK2WT mice and decreased the content of neutrophils and citH3 within the thrombi[39]. The same group demonstrated that both JAK2V617F-driven NET formation and thrombosis are dependent on PAD4; which was found overexpressed in neutrophils from patients with PV harboring JAK2V617F[39].
Finally, elegant murine studies in two different mouse models, one of them with the expression of JAK2V67F in all hematopoietic cells, and, the other one with the expression of JAK2V67F only in neutrophils, demonstrated that JAK2V617F neutrophils alone are not enough to promote NETosis and thrombosis and that they need to cooperate with platelets to induce NETs formation[43].
References
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862.
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 1–11.
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287.
- Desai, J.; Mulay, S.R.; Nakazawa, D.; Anders, H.-J. Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell. Mol. Life Sci. 2016, 73, 2211–2219.
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.-D. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (ETs) in Humans. Biomolecules 2015, 5, 702–723.
- Zawrotniak, M.; Rapala-Kozik, M. Neutrophil extracellular traps (NETs)—Formation and implications. Acta Biochim. Pol. 2013, 60, 277–284.
- Farrera, C.; Fadeel, B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J. Immunol. 2013, 191, 2647–2656.
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213.
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835.
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961.
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803.
- Steppich, B.A.; Seitz, I.; Busch, G.; Stein, A.; Ott, I. Modulation of tissue factor and tissue factor pathway inhibitor-1 by neutrophil proteases. Thromb. Haemost. 2008, 100, 1068–1075.
- Jordan, R.E.; Nelson, R.M.; Kilpatrick, J.; Newgren, J.O.; Esmon, P.C.; Fournel, M.A. Inactivation of human antithrombin by neutrophil elastase. Kinetics of the heparin-dependent reaction. J. Biol. Chem. 1989, 264, 10493–10500.
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896.
- Kambas, K.; Mitroulis, I.; Ritis, K. The emerging role of neutrophils in thrombosis—the journey of TF through NETs. Front. Immunol. 2012, 3, 1–8.
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy Mediates the Delivery of Thrombogenic Tissue Factor to Neutrophil Extracellular Traps in Human Sepsis. PLoS ONE 2012, 7, e45427.
- Rao, A.N. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases? World J. Cardiol. 2015, 7, 829.
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885.
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front. Immunol. 2017, 8.
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144.
- van Montfoort, M.L.; Stephan, F.; Lauw, M.N.; Hutten, B.A.; Van Mierlo, G.J.; Solati, S.; Middeldorp, S.; Meijers, J.C.M.; Zeerleder, S. Circulating Nucleosomes and Neutrophil Activation as Risk Factors for Deep Vein Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 147–151.
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated With Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003.
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667.
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743.
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947–956.
- Nahrendorf, M.; Swirski, F.K. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 2015, 349, 237–238.
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192.
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246.
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469.
- Hashiba, M.; Huq, A.; Tomino, A.; Hirakawa, A.; Hattori, T.; Miyabe, H.; Tsuda, M.; Takeyama, N. Neutrophil extracellular traps in patients with sepsis. J. Surg. Res. 2015, 194, 248–254.
- Ferrer-Marín, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernández-Rivas, J.M.; Hernández-Boluda, J.C.; Fernandez-Rodriguez, C.; Luño, E.; García Hernandez, C.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 34, 2648–2659.
- Olsson, A.-K.; Cedervall, J. NETosis in Cancer—Platelet–Neutrophil Crosstalk Promotes Tumor-Associated Pathology. Front. Immunol. 2016, 7, 373.
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111.
- Mauracher, L.-M.; Posch, F.; Martinod, K.; Grilz, E.; Däullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518.
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292.
- Marin Oyarzún, C.P.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Castro Ríos, M.A.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738.
- Guy, A.; Favre, S.; Labrouche-Colomer, S.; Deloison, L.; Gourdou-Latyszenok, V.; Renault, M.-A.; Riviere, E.; James, C. High circulating levels of MPO-DNA are associated with thrombosis in patients with MPN. Leukemia 2019, 33, 2544–2548.
- Kasprzycka, W.; Homa-Mlak, I.; Mlak, R.; Małecka-Massalska, T. Direct and indirect methods of evaluating the NETosis process. J. Pre-Clin. Clin. Res. 2019, 13, 50–56.
- Wolff, L.; Guy, A.; Gourdou-Latyszenokv, V.; Favre, S.; Colomer, S.; Renault, M.A.; Couffinhal, T.; James, C. Neutrophils prothrombotic characteristics during myeloproliferative neoplasms. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 205.

