 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tae Jin Kim | + 2179 word(s) | 2179 | 2021-02-09 05:21:36 | | | |
| 2 | Catherine Yang | Meta information modification | 2179 | 2021-02-20 08:43:04 | | |
Video Upload Options
Adoptive cellular approaches utilizing chimeric antigen receptor T cells (CAR-T) targeting cancer-specific antigens would be a solution for circumventing the immune tolerance mechanisms. The immunotherapeutic regimen of CAR-T cell therapy has shown potential in the eradication of hematologic malignancies, and current clinical objectives maintain the equivalent efficacy in the treatment of solid tumors, including PCa.
1. Introduction
Estimates have shown that the mortality rate of metastatic castration-resistant prostate cancer (mCRPC) and the fatal manifestation of the disease are approximately 31,000 cases annually [1]. Currently, chemotherapy with taxanes, androgen receptor axis-targeted (ARAT) agents, and immunotherapeutic approaches using sipuleucel-T and radium-223 are Food and Drug Administration-accepted therapeutic modalities that improve overall survival (OS) in men with mCRPC [2]. Significant advances and innovation in the systematic regimens and treatment protocols for mCRPC have existed since the approval of radium-223 in 2013. Immunotherapy with checkpoint inhibitors and the use of poly (ADP-ribose) polymerase inhibitors have profoundly influenced the treatment paradigm of other solid cancers [3][4]. Immunotherapy approaches targeting prostate-specific membrane antigen (PSMA), while specific to prostate cancer (PCa), may utilize the same therapeutic mechanisms relevant to other malignancies. Continued research in the field of immunotherapy will considerably alter the treatment landscape and therapeutic approaches in mCRPC and the outcomes of patients with PCa.
Recent advances in the field of immunotherapy include adoptive cellular therapy (ACT), in which the T cells from the peripheral blood are collected through leukapheresis and consequent apheresis before being genetically modified ex vivo before reinfusion [5]. Different ACT modalities, such as tumor-infiltrating lymphocytes (TILs), engineered T cell receptors (TCR), and chimeric antigen receptor T cell (CAR-T) therapy, have been engineered and are being evaluated in various scientific studies and clinical settings [5]. Clinical results from studies analyzing the effects of CAR-T therapy in solid tumors including PCa have shown promise. In the PCa treatment setting, CAR-T cells targeting PSMA have displayed encouraging results, suggesting a potential translational treatment target for the treatment of PCa in the advanced clinical setting [6]. Collectively, these clinical outcomes will open new horizons in the treatment and cure for the disease continuum of PCa. This article provides a thorough overview of the role of CAR-T cell therapy in advanced PCa and the metastatic setting will be provided along with an extensive review of current and future modalities that will further improve the therapeutic spectrum in the ongoing treatment of advanced PCa and metastatic disease.
2. The Molecular Construct of CAR-T Cells
CAR-T cells are constructed on the molecular level in which the effector portion of the T lymphocytes recognizes and attacks the cells bearing the specific surface target antigen. This interaction occurs in an environment that does not require the immunologic action of antigen-presenting cells (APC), unlike the molecular signaling required for unmodified T cell activation. Target antigen recognition and the intracellular signaling cascade in CAR-T cells are facilitated by the CAR molecule, which incorporates the recognition of the target antigen along with transmembrane and intracellular signaling. The major characteristic that distinguishes each generation of CARs is the intracellular presence of one or more costimulatory domains in the CAR. Interestingly, this variation in the costimulatory structure determines the immunologic function, molecular phenotype, and proliferative capacity of CAR-T cells and the current preferred immunotherapeutic modality in the second- or third-generation CAR layouts [7]. Moreover, CAR molecules consist of three individual components. The extracellular domain, which identifies the target antigen identification, has a single-chain fragment variable (scFv) structure that binds with tumor-associated antigens (TAA). The attachment of scFv to the T cell is possible due to the transmembrane domain, which comprises of the transmembrane regions CD 3, CD 8, CD 28, or FcεRI. The transmembrane sector is linked to the intracellular zone situated in the intracytoplasmic domains of CD 8, CD 28, CD 137, and CD 3ζ. The intracellular zone encompasses the immune receptor tyrosine-based activation motif (ITAM) that plays an essential role in signal transduction for T cell activation [8].
CARs are classified into four different classes depending on the molecular structure and complexity (Figure 1). The first-generation CAR consists of a simple receptor divided into the aforementioned components (scFv, a transmembrane domain, and intracellular zone). The first-generation CAR-T cells failed to obtain a significant number of activated T lymphocytes in blood circulation with this molecular arrangement, although enabling T cell activation was possible due to the absence of a costimulatory molecule [9][10][11]. The second-generation CAR was engineered to overcome this clinical limitation by additionally inserting a costimulatory protein (CD 28, CD 27, CD 134, or CD B7) into the intracellular domain. Additional costimulatory molecular structures such as CD 28, 4-1BB, and CD 3ζ were inserted during the development of the third-generation CAR to increase the concentration of activated T cells in the circulatory system [12]. The T cells redirected for universal cytokine-mediated killing (TRUCKs), which has a costimulatory domain and pro-inflammatory construct, such as interleukin (IL)-12, which increases the effectiveness of circulating T cells, are the fourth iteration of these molecules [13]. IL-12 aims to mitigate the immunosuppressive properties of the tumor microenvironment (TME) by activating the T helper 1-type cell cascade [14][15]. However, additional modifications to the fourth-generation CARs are not restricted to IL-12 because various categories of molecules have been engineered or modified to use in TRUCK construction. Other molecular combinations consist of cytokines, such as IL-15 to developmentally enhance the T memory stem cells [16] and IL-18 [17] along with cytokine receptors, which include the IL-7 receptor (C7R), to reduce the adverse effects of cytokine toxicity [18]. Apart from the aforementioned molecules, the knock-out genes, including PD-1 or diacylglycerol kinase, and knock-in genes, such as TCRα subunit constant gene or chemokine C-X-C motif receptor (CXCR) 4, are included in the TRUCK cassette to improve CAR expression and its antitumor properties [19][20]. Therefore, controlled and inducible systems (Syn/Notch) and antigen combinations of human epidermal growth factor receptor 2 (HER 2) + IL-13 receptor subunit α2 (IL-13Rα2) have been synthesized and utilized to mediate effective expression and prevent antigen escape [21].
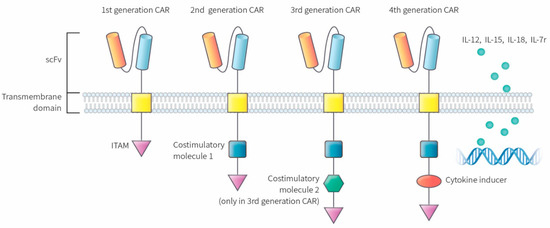
Figure 1. Molecular structure of different CAR generations. The first generation CAR only contains ITAM motifs in the intracellular domain. Second-generation CARs included the addition of one co-stimulatory molecule and third-generation CARs contain a second co-stimulatory molecule. The fourth generation of CARs was based on second-generation CAR construct paired with a constitutive cytokine inducer. These types of CAR-T cells are also known as TRUCKs. CAR: chimeric antigen receptor, scFv: single-chain fragment variable, ITAM: immunoreceptor tyrosine based activation motifs, TRUCK: T cell redirected for universal cytokine-mediated killing.
3. The CAR-T Cell Therapy Platform
Initiation of CAR-T cell therapy commences with the harvesting of lymphocytes from the peripheral blood by leukapheresis, which subsequently undergoes apheresis without the additional insertion of granulocyte colony-stimulating factor (G-CSF) [22]. The T cells are genetically refined and infected with a viral vector that can be retroviral or lentiviral in origin or non-viral CAR vector, where genomic DNA is artificially inserted. The modified CAR-T cells are introduced back into the blood circulation of the patient, who usually undergoes lymphodepletion therapy before CAR-T reinfusion, after molecular expansion and purification ex vivo [22][23]. The CAR-T cells become activated after the infused CAR interacts with the antigen presented on the surface of the target cell, which causes lysis of the target cell along with the production and proliferation of cytokines (Figure 2).
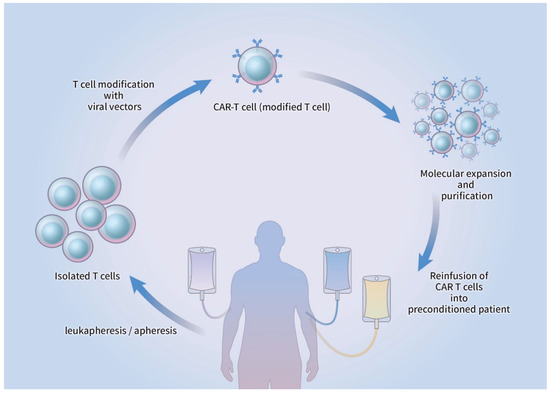
Figure 2. Schematic drawing of CAR-T cell production. T cells from peripheral blood are isolated via leukapheresis, followed by apheresis. The T cells are transduced by viral/non-viral vectors and genetically modified to express a CAR. After ex vivo expansion and purification, CAR-T cells are reinfused into the patient who received prior lymphodepletion therapy. CAR-T: chimeric antigen receptor T cell.
The molecular structure and physical properties of the receptor, transmembrane domains, and signaling zones of the individual CAR significantly influence the therapeutic effects of CAR-T cells, which make the engineering of CARs essentially an experimental approach. Moreover, CAR-T cell therapy has shown promising results in the area of hematologic malignancies, where complete response (CR) was achieved in more than half of the patients with acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and lymphoma [24][25]. The therapeutic advantages of CAR-T cell therapy are yet to be reiterated in patients with solid cancers, including breast cancer, melanoma, and sarcoma, in contrast to hematologic cancers [26][27]. The number of cancer-specific surface antigen is scarce in patients with solid cancers, and as a consequence, the success rates of the CAR-T cell therapy in destroying tumor cells and controlling TME are low. Total recognition and high identification rates of PCa-specific surface epitopes by CARs should be of paramount concern in current and novel CAR-T cell immunotherapy regimens to be effective in the treatment of metastatic PCa [28].
4. Current Limitations of CAR-T Therapy in the PCa Treatment Setting
4.1. CAR-T Cell Persistence and Tumor Trafficking
CAR-T cell therapy has opened new possibilities as an innovative modality for PCa treatment. However, studies and clinical trials based on the therapeutic efficacy of CAR-T cells have shown unsatisfactory outcomes despite highly promising preclinical results. The efficacy of the T cell function is highly dependent on its migration abilities to the target tumor and its persistence rate. The persistence rate or survival period of CAR-T cell is a major determinant in the therapeutic efficiency of these modified T cells to target and destroy tumorous lesions. Although first-generation CARs have shown a meager persistence rate, higher persistent rates have been noted in the second- and third-generation CAR-T cells [29][30].
Prolonged persistence of CAR-T cells was traditionally attained by radiotherapy or chemotherapy. Current molecular research has shown that persistence can also be achieved by the administration of oncolytic viruses, which eradicates cells with suppressive properties including myeloid and regulatory T cells. Moreover, the immunosuppressive traits of the oncolytic viruses increase tumor immunogenicity and antigenicity, leading to a higher vulnerability of tumor cells to CAR-T cells and providing an environment suitable for vascular remodeling of the tumor lesions and the proliferation of adhesion molecules [31][32][33][34].
The homing properties of CAR-T cells to tumor cells or sites are known as trafficking, a mandatory prerequisite for T cell-induced tumor eradication. CAR-T cells and natural killer (NK) cells can be modified for better migration to the tumorous lesion through the additional expression of tumor-homing receptors [35][36][37][38], manipulation of bone marrow retargeting receptors such as CXCR 4 [39][40][41], or upregulation of CXCR 3 expression, which is linked to the inhibition of protein kinase A [42]. The aforementioned methods are experimental due to the participation of multiple immune-stimulatory cascades. Therefore, further prospective clinical trials are warranted to analyze the compatibility of these regimens to be recognized as a PCa treatment modality.
4.2. The Prostate Cancer Tumor Microenvironment
The unforgiving nature of TME exhibits another challenge for effective CAR-T cell therapy. After insertion into the tumor, CAR-T cells are surrounded by a harsh TME that has a low supply of nutrients and oxygen, low pH, and compact tumor stroma with residing immunosuppressive cells that excrete inhibitory molecules and increase inflammatory reactions. Moreover, the TME has a high concentration of reactive oxygen species (ROS), which hinders the antitumor activity of CAR-T cells [31]. In addition, Ligtenberg et al. investigated the effects of catalase coexpressed CAR-T cells and concluded that the introduction of a catalase to the cells mitigated the TME oxidative state with less accretion of ROS, and the catalase expressing CAR-T cell maintained its antitumor properties [43]. Mitigating the effects or neutralizing the TME provides a molecular challenge. Therefore, numerous methods for genetically modifying CAR-T cells or altering the microenvironment have commenced or are ongoing to address this subject.
4.3. Immune-Related Adverse Events
Although adoptive immunotherapy of CAR-T cell infusion has shown the treatment benefit for PCa in various clinical trials, the majority of treatments were associated with expected and unexpected toxicities and immune-related adverse events (IRAEs), which include CRS, tumor lysis syndrome, on-target/off-tumor toxicity, and neurological side effects [44]. Furthermore, CRS has been reported as one of the predominant side effects that patients experience after CAR-T cell infusion. The activation of infused autologous CAR-T cells stimulates cytokines that cause mild to severe inflammatory reactions and can be life-threatening in some cases [45]. The clinical features of CRS include systemic (e.g., fever, malaise, fatigue, myalgia, nausea, anorexia, tachycardia, hypotension, cardiac and renal impairments, and hepatic failure) and hematologic (e.g., disseminated intravascular coagulation) symptoms [44][46]. Other possible adverse reactions caused by CAR-T cell infusion is the on-target/off-tumor recognition. The autologous infusion of activated CAR-T cell can attack normal self-cells, which can ultimately result in autoimmune disease and organ damage because the target antigen is not solely restricted to the tumor and can be expressed in other cells and organs [47]. Various molecular approaches are currently under development or evaluated to further diminish the dangers of IRAEs. A promising method of overcoming probable toxic adverse events is the genetic modification and insertion of a suicide gene into CAR-T cells to deactivate the CAR-T cell in the event of a life-threatening reaction. A clinical study done by Di Stasi et al. established the role of caspase-9 in inducing T cell apoptosis [48]. Moreover, several studies proposed the insertion of a CAR vector into NK cells, which would substantially reduce the prevalence of IRAEs [49][50]. Other experimental approaches based on hematological malignancies and solid tumors including PCa [51] suggest that the solution to reducing immunologic side effects lies in the delivery of CAR-T cells using nanoparticles [52][53].
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Pilon, D.; Ellis, L.A.; Xiao, Y.; Behl, A.S.; Lefebvre, P. Consideration of Potential Drug-Drug Interactions in Selection of FDA-Approved Drugs Indicated for Prostate Cancer. Am. J. Ther. 2019, 26, e422–e424.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461.
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004.
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18.
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431.
- Yu, H.; Pan, J.; Guo, Z.; Yang, C.; Mao, L. CART cell therapy for prostate cancer: Status and promise. OncoTargets Ther. 2019, 12, 391–395.
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944.
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115.
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971.
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365.
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706.
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277.
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89.
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581.
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141.
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247.
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144.
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266.
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69.
- Maus, M.V.; Levine, B.L. Chimeric Antigen Receptor T-Cell Therapy for the Community Oncologist. Oncologist 2016, 21, 608–617.
- Shank, B.R.; Do, B.; Sevin, A.; Chen, S.E.; Neelapu, S.S.; Horowitz, S.B. Chimeric Antigen Receptor T Cells in Hematologic Malignancies. Pharmacotherapy 2017, 37, 334–345.
- Landoni, E.; Savoldo, B. Treating hematological malignancies with cell therapy: Where are we now? Expert Opin. Biol. Ther. 2018, 18, 65–75.
- Maude, S.; Barrett, D.M. Current status of chimeric antigen receptor therapy for haematological malignancies. Br. J. Haematol. 2016, 172, 11–22.
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149.
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161.
- Lee, J.K.; Bangayan, N.J.; Chai, T.; Smith, B.A.; Pariva, T.E.; Yun, S.; Vashisht, A.; Zhang, Q.; Park, J.W.; Corey, E.; et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E4473–E4482.
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850.
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108.
- Haji-Fatahaliha, M.; Hosseini, M.; Akbarian, A.; Sadreddini, S.; Jadidi-Niaragh, F.; Yousefi, M. CAR-modified T-cell therapy for cancer: An updated review. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1339–1349.
- Lutsiak, M.E.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868.
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354.
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180.
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788.
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402.
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980.
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730.
- Arai, Y.; Choi, U.; Corsino, C.I.; Koontz, S.M.; Tajima, M.; Sweeney, C.L.; Black, M.A.; Feldman, S.A.; Dinauer, M.C.; Malech, H.L. Myeloid Conditioning with c-kit-Targeted CAR-T Cells Enables Donor Stem Cell Engraftment. Mol. Ther. 2018, 26, 1181–1197.
- Khan, A.B.; Carpenter, B.; Santos, E.S.P.; Pospori, C.; Khorshed, R.; Griffin, J.; Velica, P.; Zech, M.; Ghorashian, S.; Forrest, C.; et al. Redirection to the bone marrow improves T cell persistence and antitumor functions. J. Clin. Investig. 2018, 128, 2010–2024.
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210.
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Puré, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551.
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766.
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011.
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518.
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195.
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415.
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683.
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647.
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147.
- Jakobczyk, H.; Sciortino, F.; Chevance, S.; Gauffre, F.; Troadec, M.B. Promises and limitations of nanoparticles in the era of cell therapy: Example with CD19-targeting chimeric antigen receptor (CAR)-modified T cells. Int. J. Pharm. 2017, 532, 813–824.
- Ajina, A.; Maher, J. Prospects for combined use of oncolytic viruses and CAR T-cells. J. Immunother. Cancer 2017, 5, 90.
- Singh, S.; Asal, R.; Bhagat, S. Multifunctional antioxidant nanoliposome-mediated delivery of PTEN plasmids restore the expression of tumor suppressor protein and induce apoptosis in prostate cancer cells. J. Biomed. Mater. Res. A 2018, 106, 3152–3164.

