 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hossam Nada | + 2300 word(s) | 2300 | 2021-02-04 04:57:19 | | | |
| 2 | Lily Guo | + 71 word(s) | 2371 | 2021-02-24 11:32:29 | | |
Video Upload Options
Pathological angiogenesis is a hallmark of cancer; accordingly, a number of anticancer FDA-approved drugs act by inhibiting angiogenesis via different mechanisms. However, the development process of the most potent anti-angiogenics has met various hurdles including redundancy, multiplicity, and development of compensatory mechanisms by which blood vessels are remodeled. Moreover, identification of broad-spectrum anti-angiogenesis targets is proved to be required to enhance the efficacy of the anti-angiogenesis drugs. In this perspective, a proper understanding of the structure activity relationship (SAR) of the recent anti-angiogenics is required. Various anti-angiogenic classes have been developed over the years; among them, the heterocyclic organic compounds come to the fore as the most promising, with several drugs approved by the FDA.
1. Introduction
As the second leading cause of mortality globally, cancer has become the focus for extensive research [1][2]. Although cancer progression and metastasis consist of multiple, complex, interacting, and interdependent steps [3], angiogenesis plays an essential part of the tumor’s growth and metastasis [4], owing to the fact that growth beyond the size of l–2 mm3 requires tumors to develop an adequate blood supply [5][6]. Angiogenesis is the processes whereby new blood and lymphatic vessels form[7]. Angiogenesis and its induction remain a major hallmark of cancer as it flourishes nutrient-deprived tumors with oxygen and nutrients, thus routing tumor metastasis[8][9].
Under normal conditions, angiogenesis is essential for formation of a new vascular network to supply nutrients, oxygen, and immune cells, as well as to remove waste products [10]. This process is regulated by a balance between pro- and anti-angiogenic molecules, and once that delicate balance is disturbed[11], it could lead to various diseases, especially cancer [12]. Angiogenesis is a vital mediator of tumor development[13]. As tumors enlarge, diffusion distances from the current vascular supply rise, leading to hypoxia [14]. Continued expansion of a tumor mass needs new blood vessel formation to offer rapidly proliferating tumor cells with a suitable supply of oxygen and metabolites[15]. Without the proper blood supply, tumors cannot grow beyond a critical size or metastasize to another organ[16]. Therefore, targeting angiogenesis is one of the most effective ways to stop a tumor progression [17].
Most of the drugs that have been developed to combat angiogenesis have heterocyclics in their backbone as a common feature. This is demonstrated by heterocyclics occupying the major part of the number of FDA-approved anti-angiogenic drugs, as demonstrated in Figure 1. This domination of FDA-approved drugs is likely due to the wide availability of various heterocyclic fragments that have different potency, physicochemical properties, and lipophilicity. This versatility makes heterocyclic fragments a sound rational choice when developing new drugs or altering an existing one.
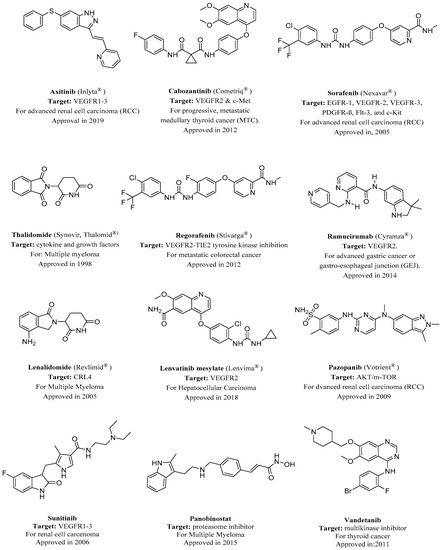
Figure 1. FDA-approved angiogenesis inhibitors for treatment of cancer[18][19]. VEGFR, vascular endothelial growth factor receptor.
2. Nitrogen-Based Heterocycles
The number of anti-cancer candidates possessing a nitrogen heterocycle is an indicator of the structural significance of nitrogen-based heterocycles in the fight against cancer [20][21][22]. More than 75% of drugs approved by the FDA and currently available in the market are nitrogen-containing heterocyclic moieties [23]. One of the most important targets in angiogenesis inhibition is vascular endothelial growth factor receptors (VEGFRs).
Vascular endothelial growth factor is an important signaling protein involved in both vasculogenesis (the formation of the circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature)[24][25]. As its name implies, VEGF activity is restricted mainly to cells of the vascular endothelium [26]. The expression of VEGF is potentiated in response to hypoxia, by activated oncogenes, and by a variety of cytokines [27]. VEGF induces endothelial cell proliferation, promotes cell migration, and inhibits apoptosis [28]. VEGF induces angiogenesis as well as permeabilization of blood vessels, and plays a central role in the regulation of vasculogenesis[29][30].
Deregulated VEGF expression contributes to the development of solid tumors by promoting tumor angiogenesis and to the etiology of several additional diseases characterized by abnormal angiogenesis[31]. Consequently, inhibition of VEGF signaling abrogates the development of a wide variety of tumors[32]. All members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors (the VEGFRs) on the cell surface, causing them to dimerize and become activated through transphosphorylation[33][34].
Several nitrogen-based heterocyclic drugs have been used to inhibit VEGF, for example, agents that inhibit the VEGFR tyrosine kinase such as the pyrrolidinone-based Sunitinib were approved by the FDA for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor[35][36]. Another example is the phthalazine-based vatalanib, which is under investigation for the treatment of metastatic colorectal cancer and non-small cell lung cancer (NSCLC) [37][38]. Indole ring is one of the nitrogen-based heterocyclics that have been involved in VEGF inhibition[39]. It has demonstrated the ability to inhibit the proliferation, growth, and invasion of human cancer cells. Panobinostat is an indole-based drug approved in 2015 for the treatment of multiple myeloma[40].
Renhowe et al. synthesized a series of quinolin-2-one analogues that showed promising VEGFR-2 inhibition activity (Table 1)[41]. Upon examining the series of analogues synthesized by Renhowe et al., it was found that the free NH of the hydroquinolin-2-one scaffold (Figure 2), the quinolinone carbonyl, and the benzimidazole NH formed donor–acceptor motifs that would bind to the hinge region of the VEGFR-2, which could be implicated in tumor vasculature formation and maintenance [42][43]. The importance of a hydrogen donating group was found through substituting different groups at the C4 position, observing the potency increasing with (NH2 > OH > H). The incorporation of large basic amine at the C4 position such as an amino quinuclidine resulted in much more potent activity; however, it negatively affected the pharmacokinetics. Therefore, the NH2 substitution at C4 was maintained and subsequent efforts focused on modifying the substitution on both rings A and D to attain additional enzymatic affinity and to improve physicochemical properties.
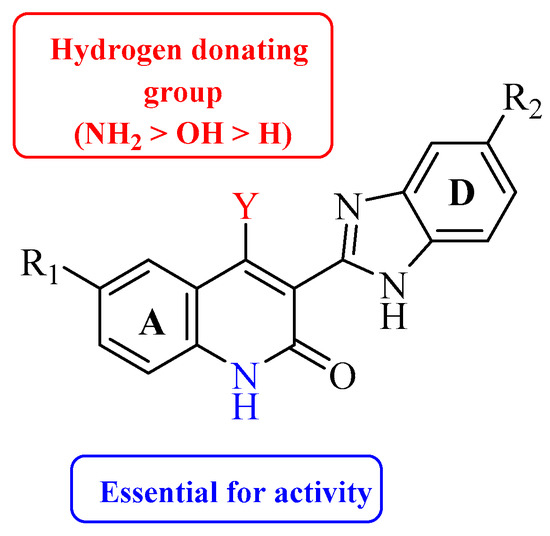
Figure 2. Structure–activity relationship study of 3-benzimidazol-2- ylhydroquinolin-2-one derivatives.
Table 1. In vitro inhibition of vascular endothelial growth factor receptor 2 (VEGFR-2) by the quinolin-2-one analogues (1–10) [41].
| Compound | Y | R1 | R2 | VEGFR-2 (IC50 μM) | PDGFRβ (IC50 μM) |
|---|---|---|---|---|---|
| 1 | OH | H | H | 0.24 | 0.020 |
| 2 | NH2 | H | H | 0.058 | 0.010 |
| 3 | NHMe | H | H | 0.22 | 0.030 |
| 4 | 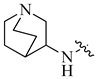 |
H | H | 0.17 | 0.0002 |
| 5 | H | 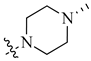 |
H | 0.017 | 0.0003 |
| 6 | NH2 | 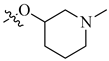 |
H | 0.005 | 0.0001 |
| 7 | NH2 | H | Me | 0.22 | 0.030 |
| 8 | NH2 | H | 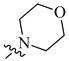 |
0.057 | 0.005 |
| 9 | NH2 | H | 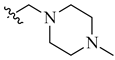 |
0.027 | 0.002 |
| 10 | NH2 | H | 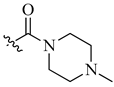 |
0.026 | 0.0009 |
As illustrated in Figure 3, the interaction of compound 10 with VEGFR-2 receptor active site (IC50 = 0.026 μM) was subjected to docking studies using Schrodinger maestro. The modelling simulation shows a significant docking score of −9.058, along with the formation of two hydrogens bonds as well as two π–cation bonds. These interactions were found to be energetically significant as all the formed bonds among the donor and acceptor atoms are within about 3.7 Å.
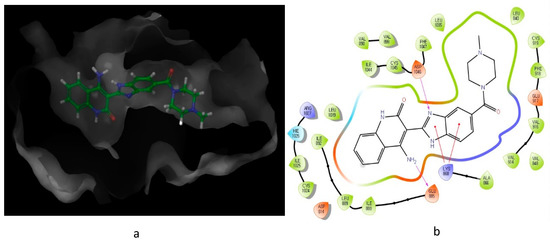
Figure 3. Predicted interaction of compound 10 (imidazole-scaffold) with active site residues of VEGFR-2. (a) 3D structural view of ligand inside the receptor’s active site. (b) Ligand interaction diagram; violet lines represent hydrogen bonds and red lines represent π–cation interactions.
Although VEGF is an important pathway in angiogenesis, several other targets are also involved in angiogenesis. Matrix metalloproteinases (MMPs) are one such target involved in angiogenesis[44]. MMPs are implicated in early steps of tumor evolution including stimulation of cell proliferation and modulation of angiogenesis[45]. During angiogenesis, new vessels develop from present endothelial lined vessels to encourage the degradation of the vascular basement membrane and remodel the extracellular matrix (ECM), followed by endothelial cell migration, proliferation, and formation of the new generation of matrix components[46][47].
Matrix metalloproteinases participate in the disruption, tumor neovascularization, and successive metastasis, while tissue inhibitors of metalloproteinases (TIMPs) downregulate the activity of these MMPs [48]. Therefore, MMPs, through the modulation of the balance between pro- and anti-angiogenic factors, can directly or indirectly mediate the angiogenic response [49].
The relation between MMP overexpression in tumor and cancer progression has encouraged the progress of preclinical trials with a series of inhibitors designed to block the proteolytic activity of these enzymes.
Thus far, no FDA-approved MMP inhibitors for the treatment of cancer have emerged. However, several leads have been developed; for example, Becker et al. reported new piperidine α-sulfone hydroxamates with potent matrix metalloproteinase inhibition activity[50]. The α- and β-sulfone derivatives showed a difference in the inhibitory activity toward the metalloproteinases. The β-sulfone hydroxamate showed potency for the targeted MMPs and selectivity for MMP-1, but generally exhibited poor oral bioavailability. In addition, some β-sulfones with α-hydrogens can undergo β-elimination. On the other hand, the α-sulfones possess both potency and selectivity and provide an improvement in oral exposure demonstrated by a higher Cmax value and bioavailability relative to the β-sulfones.
On further investigation, they concluded that α-sulfone derivatives are two to four times more potent and bioavailable than the β-sulfones. The higher bioavailability and Cmax may be due to greater steric bulk around the hydroxamate, protecting it from the usual modes of hydroxamate metabolism including N-O bond cleavage, hydrolysis, and glucuronidation. The findings of Becker et al. can be explained through the comparison of the different activities of α- and β-sulfone derivatives, as demonstrated in Table 2.
Table 2. Comparing α- and β-sulfone derivatives for their matrix metalloproteinase 1 (MMP-1) activity [50].
 |
||||||
| Ki (nM) | ||||||
| Compound | MMP-1 | MMP-2 | MMP-3 | MMP-9 | MMP-13 | Cmax |
| 11 (β-sulfone) | 800 | 0.4 | 17.5 | 1 | 0.6 | 1372 |
| 12 (α-sulfone) | 435 | ˂0.1 | 18.1 | 0.3 | 0.15 | 3119 |
Looking at the interaction of the α- and β-sulfone derivatives (Figure 4 and Figure 5) with the active site residues of MMP-2 [51], the difference in potency can be elucidated because of the fact that the α-derivative showed a docking score of −7.054, while its counterpart, the β-derivative, exhibited a docking score of −5.22. This difference in the docking scores as well as forming different interactions with the active site could explain why the α-sulfone derivative would be a more promising lead for further development.
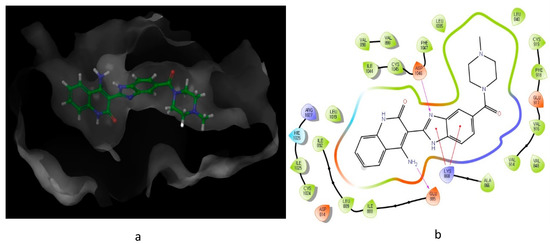
Figure 4. Predicted interaction of α-sulfone (12) derivative active site residues of matrix metalloproteinase (MMP) [51,52]. (a) 3D structural view of ligand inside the receptor’s active site. (b) Ligand interaction diagram; violet lines represent hydrogen bonds and green lines and grey line represent metal interactions.
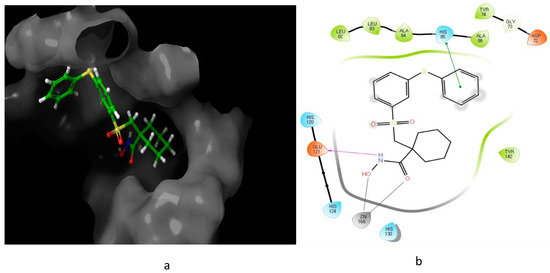
Figure 5. Predicted interaction of β-sulfone (11) derivative active site residues of MMP. (a) 3D structural view of ligand inside the receptor’s active site. (b) Ligand interaction diagram; violet lines represent hydrogen bonds, green lines represent π–π interactions, and grey line represent metal interactions.
3. Heterocyclic Sulfonamides
Sulfonamides significance stems from the fact that they constitute a significant class of drugs with various types of pharmacological effects including antitumor, anti-carbonic anhydrase, and diuretic activity[53][55][56][54]. There are many reports of a multitude of structurally novel heterocyclic sulfonamide derivatives that have been stated to display significant antitumor activity[57][58]. Although the mechanism by which they combat cancer may vary, the majority of anti-cancer sulfonamides act on angiogenesis through several pathways.
As of today, there are no FDA-approved heterocyclic sulfonamide anti-angiogenics in the market, but there are several promising leads being developed. One of such leads is E7820, which is now in phase II clinical trial for colorectal cancer treatment through the inhibition of integrin α2 [59]. Integrins are transmembrane receptors that are central to the biology of many human pathologies, where they act through the mediation of the cell-extracellular matrix and cell–cell interaction [60]. This mediative action is carried out through the transduction of information from the extracellular environment to modulate cell responses, together with adhesion, spreading, migration, growth signaling, survival signaling, secretion of proteases, and invasion[3][61][62]. Numerous studies report that the increased levels of integrin α2 facilitate the spread of cancer [63][64][65][66]. Thus, inhibitors of integrin α2, such as that shown in Figure 6, will slow the spread of cancer and its metastasis.
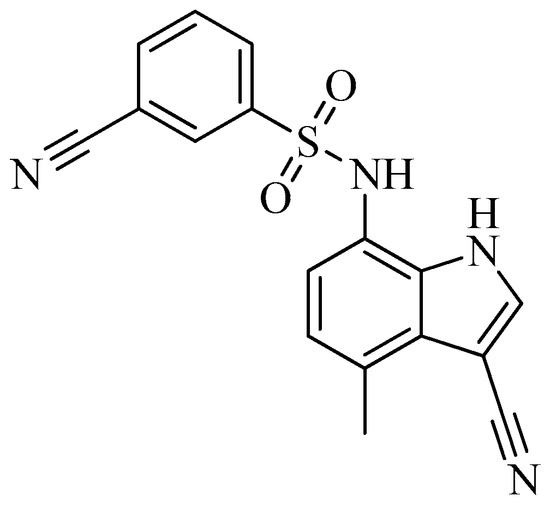
Figure 6. Chemical structure of the heterocyclic sulfonamide anti-angiogenic E7820.
Hypoxia inducible factors (HIFs) are another promising anti-angiogenic inhibitor[67]. This is because of the requirement of an adequate oxygen supply for the macroscopic tumor to grow [68]. This need is fulfilled through tumor angiogenesis, which results from an increased synthesis of angiogenic factors and a decreased synthesis of anti-angiogenic factors[49][69].
The shift of the balance between pro- and anti-apoptotic factors due to the metabolic adaptation of tumor cells to decrease the oxygen availability by increasing glucose transport and glycolysis to promote survival is prominent in hypoxia[69][70]. In this regard, HIF-1, which is induced by many factors, is mainly implicated in tumor angiogenesis.
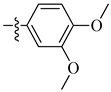
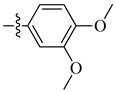
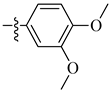
A class of heterocyclics based on the lead sulfonamide hits identified by Gerguson et al. showed potent activity against hypoxia (Table 3) [71][72]. It was found that the oxygen atom in ring A was essential for the activity, while the double bond in the same ring resulted in increased hepatotoxicity, leading to the conclusion that the double bond should be removed. Furthermore, the sulfonamide moiety as well as the hydrophobic substitutions in ring B increased the activity significantly. In ring C, the methoxy substituent at para position is essential for activity, while meta methoxy is not. Additionally, upon replacing the para methoxy with electron withdrawing groups, the activity was lost, demonstrating that only electron donating groups should occupy this position. This SAR is summarized in Figure 7.
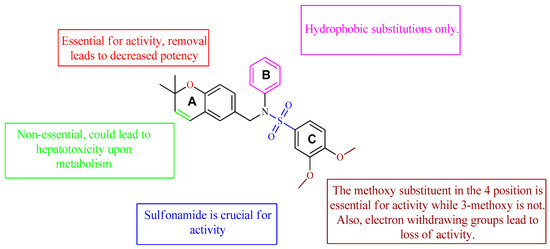
Figure 7. Structure–activity relationship study of the benzenesulfonamide hits and its analogues [71][72].
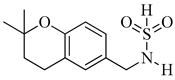
Table 3. In vitro inhibition of hypoxia inducible factor 1 (HIF-1) transcriptional activity in cell-based HRE reporter assay by the benzenesulfonamide analogues (13–18) [72].
 |
|||
| Compound | R1 | R2 | LN229-HRE-Lux (IC50 μM) |
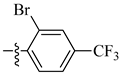
| 13 | 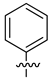 |
 |
0.6 μM |
| 14 | 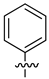 |
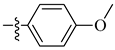 |
0.6 μM |
| 15 | 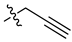 |
 |
1.3 μM |
| 16 | 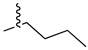 |
 |
3.3 μM |
| 17 | 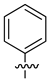 |
 |
>25 μM |
| 18 | 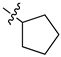 |
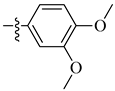 |
0.5 μM |
The interaction of compound 13, possessing IC50 of 0.6 μM, with HIF-1 active site residue, is demonstrated in Figure 8. It showed better binding energy and inhibition constant as compared with N-((2,2-dimethylchroman-6-yl) methyl) sulfonic amide, with docking scores of −6.85 and −5.4, respectively. This could be owing to the ability of the sulfonamide derivative (13) to form a hydrogen bond and metal interaction with the active site residue.
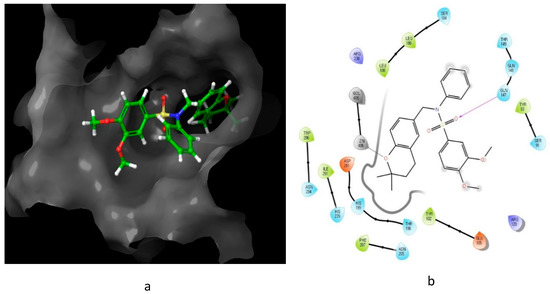
Figure 8. Predicted interaction of compound 13 with hypoxia-inducible factor active site residue. (a) 3D structural view of the sulfonamide ligand inside the receptor’s active site. (b) Ligand interaction diagram; violet lines represent hydrogen bonds and grey lines represent metal interaction.
4. Conclusions
Throughout this review, several leads were investigated for SAR, molecular interaction, and in silico pharmacokinetics study. The following outlines were concluded, aiming to develop more active analogues of the discussed leads. Compound 10 and its derivatives would benefit from both a modification to prevent its efflux by the PGP as well as modifications to increase its interaction with the VEGFR-2 active site. Both compounds 12 and 13 and their derivatives show good drug-likeness properties, but would benefit from further isosteric changes to their structures to increase their interactions with their respective active site. Paclitaxel and its derivatives have shown the highest binding potential to MTA active site residue, with docking scores around −9.5. However, they all violated Lipinski’s rule of five, leading to poor bioavailability. Therefore, further modification to their chemical structure is highly needed.
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D.J. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90.
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet‐Tieulent, J.; Jemal, A.J. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108.
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Drug Discov. 2018, 17, 31–46.
- Mashreghi, M.; Azarpara, H.; Bazaz, M.R.; Jaafari, M.R.; Masoudifar, A.; Mirzaei, H.; Jaafari, M.R. Angiogenesis biomarkers and their targeting ligands as potential targets for tumor angiogenesis. J. Cell. Physiol. 2018, 233, 2949–2965, doi:10.1002/jcp.26049.
- Ellis, L.; Fidler, I. Angiogenesis and metastasis. Eur. J. Cancer 1996, 32, 2451–2460, doi:10.1016/s0959-8049(96)00389-9.
- Schättler, H.; Ledzewicz, U. Optimal Control for Mathematical Models of Cancer Therapies; Springer: Berlin/Heidelberg, Germany, 2015; Volume 42.
- McColl, B.K.; Stacker, S.A.; Achen, M.G. Molecular regulation of the VEGF family–inducers of angiogenesis and lymphangiogenesis. J. Pathol. Microbiol. Immunol. 2004, 112, 463–480.
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281, doi:10.1016/j.semcancer.2018.10.002.
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-Mediated Angiogenesis Links EMT-Induced Cancer Stemness to Tumor Initiation. Cancer Res. 2014, 74, 1566–1575, doi:10.1158/0008-5472.can-13-1641.
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514, doi:10.1038/nrc2868.
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells, Mol. Dis. 2007, 39, 212–220, doi:10.1016/j.bcmd.2007.04.001.
- Fidler, I.J. Angiogenesis and cancer metastasis. Cancer J. 2000, 6, S134–S141.
- Nussenbaum, F.; Herman, I.M. Tumor Angiogenesis: Insights and Innovations. J. Oncol. 2010, 2010, 1–24, doi:10.1155/2010/132641.
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750, doi:10.1038/nrc.2017.93.
- Ronca, R.; Benkheil, M.; Mitola, S.; Struyf, S.; Liekens, S. Tumor angiogenesis revisited: Regulators and clinical implications. Med. Res. Rev. 2017, 37, 1231–1274, doi:10.1002/med.21452.
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; Dubois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665, doi:10.1158/0008-5472.can-16-3199.
- Miyake, M.; Goodison, S.; Lawton, A.; Gomes-Giacoia, E.; Rosser, C. Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene 2015, 34, 890–901.
- Al-Abd, A.M.; AlAmoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605, doi:10.1016/j.jare.2017.06.006.
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171, doi:10.1159/000488087.
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Yar, M.S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189, doi:10.1016/j.ejmech.2016.09.023.
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198, doi:10.1016/j.ejmech.2018.03.055.
- Elkamhawy, A.; Park, J.-E.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 158, doi:10.3109/14756366.2015.1004057.
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909, doi:10.3390/molecules25081909.
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016). Expert Opin. Ther. Patents 2017, 27, 987–1004, doi:10.1080/13543776.2017.1344215.
- Cheng, K.; Liu, C.-F.; Rao, G.-W. Anti-angiogenic Agents: A Review on Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) Inhibitors. Curr. Med. Chem. 2020, doi:10.2174/0929867327666200514082425.
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8, doi:10.1161/hypertensionaha.117.10271.
- Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Zelli, V.; Guadagni, S.; Mackay, A.R. Hypoxia-induced alternative splicing: The 11th Hallmark of Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–30, doi:10.1186/s13046-020-01616-9.
- Yan, X.; Hui, Y.; Hua, Y.; Huang, L.; Wang, L.; Peng, F.; Tang, C.; Liu, D.; Song, J.; Wang, F. EG-VEGF silencing inhibits cell proliferation and promotes cell apoptosis in pancreatic carcinoma via PI3K/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2019, 109, 762–769, doi:10.1016/j.biopha.2018.10.125.
- Schafer, C.M.; Gurley, J.M.; Kurylowicz, K.; Lin, P.K.; Chen, W.; Elliott, M.H.; Davis, G.E.; Bhatti, F.; Griffin, C.T. An inhibitor of endothelial ETS transcription factors promotes physiologic and therapeutic vessel regression. Proc. Natl. Acad. Sci. USA 2020, 117, 26494–26502.
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22, doi:10.1096/fasebj.13.1.9.
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426, doi:10.1007/s10456-017-9562-9.
- Rosen, L.S. Clinical Experience with Angiogenesis Signaling Inhibitors: Focus on Vascular Endothelial Growth Factor (VEGF) Blockers. Cancer Control. 2002, 9, 36–44, doi:10.1177/107327480200902s05.
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225, doi:10.1016/s0092-8674(00)00114-8.
- Park, S.A.; Jeong, M.S.; Ha, K.-T.; Jang, S.B. Structure and function of vascular endothelial growth factor and its receptor system. BMB Rep. 2018, 51, 73–78, doi:10.5483/bmbrep.2018.51.2.233.
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; De Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731, doi:10.1056/nejmoa1303989.
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359, doi:10.1200/jco.2007.15.7461.
- Scagliotti, G.V.; Govindan, R. Targeting Angiogenesis with Multitargeted Tyrosine Kinase Inhibitors in the Treatment of Non‐Small Cell Lung Cancer. Oncology 2010, 15, 436–446, doi:10.1634/theoncologist.2009-0225.
- Lin, Z.; Zhang, Q.; Luo, W. Angiogenesis inhibitors as therapeutic agents in cancer: Challenges and future directions. Eur. J. Pharmacol. 2016, 793, 76–81, doi:10.1016/j.ejphar.2016.10.039.
- Liu, J.-C.; Narva, S.; Zhou, K.; Zhang, W. A Review on the Antitumor Activity of Various Nitrogenous-based Heterocyclic Compounds as NSCLC Inhibitors. Mini Rev. Med. Chem. 2019, 19, 1517–1530, doi:10.2174/1389557519666190312152358.
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704.
- Renhowe, P.A.; Pecchi, S.; Shafer, C.M.; Machajewski, T.D.; Jazan, E.M.; Taylor, C.; Antonios-McCrea, W.; McBride, C.M.; Frazier, K.; Wiesmann, M.; et al. Design, Structure−Activity Relationships and in Vivo Characterization of 4-Amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A Novel Class of Receptor Tyrosine Kinase Inhibitors. J. Med. Chem. 2009, 52, 278–292, doi:10.1021/jm800790t.
- Kubo, H.; Fujiwara, T.; Jussila, L.; Hashi, H.; Ogawa, M.; Shimizu, K.; Awane, M.; Sakai, Y.; Takabayashi, A.; Alitalo, K.J.B. Involvement of vascular endothelial growth factor receptor-3 in maintenance of integrity of endothelial cell lining during tumor angiogenesis. J. Am. Soc. Hematol. 2000, 96, 546–553.
- McMahon, G.VEGF receptor signaling in tumor angiogenesis. Oncologist 2000, 5, 3–10.
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241.
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27, doi:10.1111/j.1742-4658.2010.07919.x.
- Spuul, P.; Daubon, T.; Pitter, B.; Alonso, F.; Fremaux, I.; Kramer, I.; Montañez, E.; Génot, E. VEGF-A/Notch-Induced Podosomes Proteolyse Basement Membrane Collagen-IV during Retinal Sprouting Angiogenesis. Cell Rep. 2016, 17, 484–500, doi:10.1016/j.celrep.2016.09.016.
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801, doi:10.1038/nrm3904.
- González-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. 2019, 137, 57–83, doi:10.1016/j.critrevonc.2019.02.010.
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337, doi:10.1016/j.semcancer.2009.05.003.
- Becker, D.P.; Villamil, C.I.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; DeCrescenzo, G.A.; Freskos, J.N.; Getman, D.P.; Hockerman, S.; Heintz, R.J.; et al. Synthesis and Structure−Activity Relationships of β- and α-Piperidine Sulfone Hydroxamic Acid Matrix Metalloproteinase Inhibitors with Oral Antitumor Efficacy. J. Med. Chem. 2005, 48, 6713–6730, doi:10.1021/jm0500875.
- Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. http://www.rcsb.org/structure/1HOV (accessed on 2020/11/1).
- Feng, Y.; Likos, J. J.; Zhu, L.; Woodward, H.; Munie, G.; McDonald, J. J.; Stevens, A. M.; Howard, C. P.; De Crescenzo, G. A.; Welsch, D.; Shieh, H. S.; Stallings, W. C., Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochimica et biophysica acta 2002, 1598, (1-2), 10-23, doi: 10.1016/S0167-4838(02)00307-2.
- Gulçin, I.; Taslimi, P. Sulfonamide inhibitors: A patent review 2013-present. Expert Opin. Ther. Patents 2018, 28, 541–549, doi:10.1080/13543776.2018.1487400.
- Awadallah, F.M.; Bua, S.; Mahmoud, W.R.; Nada, H.H.; Nocentini, A.; Supuran, C.T. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J. Enzym. Inhib. Med. Chem. 2018, 33, 629–638, doi:10.1080/14756366.2018.1446432.
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Bua, S.; Lee, S.H.; AbdelGawad, M.A.; Roh, E.J.; Eldehna, W.M.; Supuran, C.T. Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino)benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1457–1464, doi:10.1080/14756366.2019.1652282.
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Lee, K.; El Kerdawy, A.M.; Abbas, B.S.N.; Roh, E.J.; Eldehna, W.M.; Elshemy, H.A.; Bakr, R.B.; et al. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorganic Med. Chem. 2020, 28, 115525, doi:10.1016/j.bmc.2020.115525.
- Owa, T.; Nagasu, T. Novel sulphonamide derivatives for the treatment of cancer. Expert Opin. Ther. Patents 2000, 10, 1725–1740, doi:10.1517/13543776.10.11.1725.
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 671–686, doi:10.1080/17460441.2020.1743676.
- Funahashi, Y.; Sugi, N.H.; Semba, T.; Yamamoto, Y.; Hamaoka, S.; Tsukahara-Tamai, N.; Ozawa, Y.; Tsuruoka, A.; Nara, K.; Takahashi, K. Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res. 2002, 62, 6116–6123.
- Pan, L.; Zhao, Y.; Yuan, Z.; Qin, G. Research advances on structure and biological functions of integrins. SpringerPlus 2016, 5, 1–11, doi:10.1186/s40064-016-2502-0.
- Hytönen, V.P.; Wehrle-Haller, B. Mechanosensing in cell–matrix adhesions–Converting tension into chemical signals. Exp. Cell Res. 2016, 343, 35–41.
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473, doi:10.1038/s41580-019-0134-2.
- Mizejewski, G.J. Role of integrins in cancer: Survey of expression patterns. Proc. Soc. Exp. Boil. Med. 1999, 222, 124–138.
- Lu, X.; Lu, D.; Scully, M.; Kakkar, V. The Role of Integrins in Cancer and the Development of Anti-Integrin Therapeutic Agents for Cancer Therapy. Perspect. Med. Chem. 2008, 2, 57–73, doi:10.1177/1177391x0800200003.
- Yousefi, H.; Vatanmakanian, M.; Mahdiannasser, M.; Mashouri, L.; Alahari, N.V.; Monjezi, M.R.; Ilbeigi, S.; Alahari, S.K. Understanding the role of integrins in breast cancer invasion, metastasis, angiogenesis, and drug resistance. Oncogene 2021, 1–21, doi:10.1038/s41388-020-01588-2.
- Yoshimura, K.; Meckel, K.F.; Laird, L.S.; Chia, C.Y.; Park, J.-J.; Olino, K.L.; Tsunedomi, R.; Harada, T.; Iizuka, N.; Hazama, S.; et al. Integrin α2 Mediates Selective Metastasis to the Liver. Cancer Res. 2009, 69, 7320–7328, doi:10.1158/0008-5472.can-09-0315.
- Tan, C.; De Noronha, R.G.; Devi, N.S.; Jabbar, A.A.; Kaluz, S.; Liu, Y.; Mooring, S.R.; Nicolaou, K.; Wang, B.; Van Meir, E.G. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5528–5532, doi:10.1016/j.bmcl.2011.06.099.
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ’Go or Grow’: The key to the emergence of invasion in tumour progression?. Math. Med. Biol. 2012, 29, 49–65, doi:10.1093/imammb/dqq011.
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164, doi:10.1016/j.canlet.2014.04.001.
- Bacon, A.; Harris, A.L. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004, 36, 530–539, doi:10.1080/07853890410018231.
- Ferguson, J.; De Los Santos, Z.; Devi, N.; Van Meir, E.; Zingales, S.K.; Wang, B. Examining the structure-activity relationship of benzopyran-based inhibitors of the hypoxia inducible factor-1 pathway. Bioorg. Med. Chem. Lett. 2017, 27, 1731–1736, doi:10.1016/j.bmcl.2017.02.073.
- Bhattarai, D.; Xu, X.; Lee, K. Hypoxia‐inducible factor‐1 (HIF‐1) inhibitors from the last decade (2007 to 2016): A “structure–activity relationship” perspective. Med. Res. Rev. 2018, 38, 1404–1442.


