 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chathura Suraweera | + 4926 word(s) | 4926 | 2021-01-29 04:14:13 | | | |
| 2 | Marc Kvansakul | -1 word(s) | 4925 | 2021-02-15 00:45:54 | | | | |
| 3 | Peter Tang | Meta information modification | 4925 | 2021-02-15 13:04:13 | | |
Video Upload Options
Apoptosis is a form of cellular suicide initiated either via extracellular (extrinsic apoptosis) or intracellular (intrinsic apoptosis) cues. This form of programmed cell death plays a crucial role in development and tissue homeostasis in multicellular organisms and its dysregulation is an underlying cause for many diseases. Intrinsic apoptosis is regulated by members of the evolutionarily conserved B-cell lymphoma-2 (Bcl-2) family, a family that consists of pro- and anti-apoptotic members. Bcl-2 genes have also been assimilated by numerous viruses including pox viruses, in particular the sub-family of chordopoxviridae, a group of viruses known to infect almost all vertebrates. The viral Bcl-2 proteins are virulence factors and aid the evasion of host immune defenses by mimicking the activity of their cellular counterparts. Viral Bcl-2 genes have proved essential for the survival of virus infected cells and structural studies have shown that though they often share very little sequence identity with their cellular counterparts, they have near-identical 3D structures.
1. Introduction
Apoptosis is a form of programmed cell death activated in response to either extracellular (extrinsic apoptosis) or intracellular (intrinsic apoptosis) stimuli [1]. Playing a pivotal role in development and tissue homeostasis in multicellular organisms apoptosis selectively removes unwanted, damaged, or pathogen infected cells [2]. Apoptosis probably arose initially as a defense mechanism against pathogens and was subsequently adapted for additional purposes such as control of tissue morphogenesis during development [3][4]. While necessary for homeostasis and other regulatory roles, subversion of apoptosis underlies an array of diseases including cancers and autoimmune diseases [5]. The importance of regulating host cell death and immune responses has triggered the assimilation of many apoptotic regulatory genes by viruses. Poxviruses, in particular, have captured numerous genes for manipulating apoptosis such as viral Bcl-2 (vBcl-2) homologs, serpin protease inhibitors, dsRNA inhibitors, NF-κB inhibitors [6][7], or Interferon (IFN) inhibitors [8][9][10].
Metazoans share homologous genes that regulate both intrinsic and extrinsic apoptosis such as Bcl-2 proteins, caspases, and adaptor proteins [4]. Both intrinsic and extrinsic apoptosis initiation are governed by the activation of cysteine aspartyl proteases (caspases) that degrade intracellular targets [11]. However, there are significant differences in how the caspase cascade is initiated in intrinsic apoptosis compared with extrinsic apoptosis.
Extrinsic or death receptor mediated apoptosis initiates when death ligand (Fas L/TNF-α)/TNF related apoptosis inducing ligands (TRAIL) bind and oligomerize tumor necrosis factor (TNF) superfamily receptors at the cell surface. The TNF superfamily receptors bear cytoplasmic death domains (DD) as protein interaction modules [12] that recruit intracellular ligands through protein–protein interactions between receptor DDs and ligand DDs. TNF-receptor superfamily signaling is complex and the resultant biological outcome is dependent on the extracellular ligand bound and activates both apoptotic and non-apoptotic pathways [13]. Adaptor proteins such as FADD (Fas associated death domain protein) or TRADD (TNF receptor associated death domain protein) are recruited via their DDs once TNF receptors are activated to form the multiprotein death inducing signaling complex (DISC). DISC formation is essential for downstream activation of the caspase cascade [12][14] and there are numerous post-translational controls on their activation [15]. At the DISC, inactive pro-caspase-8 undergoes proteolytic cleavage to release p18/p12 domain to form the active caspase-8 homodimer that subsequently proteolytically activates the executioner caspases (caspase-3, -6, and -7). In addition to activating the caspases, active caspase-8 cleaves the cellular BH3 interacting domain (Bid) protein to activate this BH3-only protein for Bcl-2 initiated apoptosis and thus links extrinsic apoptosis with intrinsic apoptosis [17].
Intrinsic apoptosis, in contrast to extrinsic apoptosis, is initiated by intracellular signals and is primarily regulated by Bcl-2 family genes [1][5]. Though not all metazoans share the Bcl-2 genes [19], they generally have been well conserved from the earliest metazoans such as sponges, placozoans, and cnidarians [19][20][21][22] to worms [23], fish [24], and humans [25]. The Bcl-2 family is split into pro- and anti-apoptotic Bcl-2 members that all share up to four conserved sequence regions or Bcl-2 homology motifs (BH1-BH4), and are central to their action [19][26]. The pro-apoptotic Bcl-2 proteins are further subdivided into two groups, the multi-motif pro-apoptotic Bcl-2 (whose members include Bax, Bak, and Bok) that are closely related in sequence and structure to the pro-survival Bcl-2 proteins, and the BH3-only proteins (Bad, Bid, Bim, Bik, Bmf, Hrk, Noxa, and Puma) that are phylogenomically more distant [27]. Several models have been proposed for the action of the BH3-only proteins, but their main role appears to be to inhibit the pro-survival Bcl-2 proteins, though they may also activate some pro-apoptotic members [28]. Structural studies have shown that the BH3 region of pro-apoptotic proteins binds in a groove provided by the pro-survival protein [27]. Notwithstanding the unresolved issue regarding interaction with Bax or Bak, BH3-only proteins are major apoptosis inducers that are activated in response to various cellular insults and initiate the cell death process, leading to Bak and Bax oligomerization at the mitochondria outer membrane (MOM) and its permeabilization (MOMP) [29]. MOMP leads to the release of cytochrome c and other apoptosis inducing factors from the mitochondria inter membrane space that activates the caspase cascade that ultimately leads to cell death [30]. Other roles have also been ascribed to the Bcl-2 proteins including autophagy [31][32] and cytosolic Ca2+ regulation [33].
Since apoptosis plays an important front-line defense mechanism against invading pathogens [1][26], viruses have evolved multiple strategies to block host cell apoptosis [7][34] to enable their successful infection and replication [35]. Most large DNA viruses including the poxviruses utilize protective responses during infection to keep the host-cells alive by molecular mimicry, allowing them to produce structural, functional, and sequence homologs of cellular pro-survival proteins including Bcl-2 proteins to overturn host cell apoptosis [7][34]. Pro-survival genes including those of the Bcl-2 family have been acquired by viruses [2][19]. While Bcl-2 genes are not the only pro-survival factors in viral genomes, they are probably the most well understood at a molecular level [36]. The pro-survival Bcl‑2 proteins exist as a globular helical bundle comprising seven or eight alpha helices, with helices α2-α5 forming the canonical hydrophobic ligand binding groove that provides the interaction site for BH3 motifs of pro-apoptotic Bcl-2 proteins [2]. Not all Bcl-2 mimics share significant sequence similarity with mammalian Bcl-2 proteins, indeed, some have very low shared identity (<10%), which makes them difficult to identify from their sequence alone [37][38][39]. For example, the Bcl-2 homolog of Epstein Barr Virus (EBV) BHRF1 has 17.5% shared identity with human Bcl-xL with homologous residues clustering in BH motifs, whereas the myxoma virus homolog M11L shares only 9.6% and features no obvious BH motifs [37], however, both vBcl-2 homologs adopt the canonical Bcl-2 fold, obstruct premature host cell death, and are critical for successful infection and proliferation [40].
Subversion of host cell apoptosis by viral Bcl-2 homologs employs the interactions between pro-survival vBcl-2 proteins and cellular pro-apoptotic Bcl-2 proteins Bax, Bak, or BH3-only proteins, in a similar mode of action of their cellular counterparts [41] (Figure 1). These interactions have been widely studied and affinity measurements reported and the major finding is that there are significant variations in the BH3 binding profile of the vBcl-2 proteins [41]. The structures of a number of vBcl-2 proteins either ligand free or as complexes with their potential cellular targets were determined (Figure 1). These findings suggested that some viruses need apoptosis to escape the host cell and thus apoptosis is only delayed, not prevented. Other viruses need to block the apoptosis by selectively mimicking and supplementing the action of particular pro-survival Bcl-2 proteins, which are important during viral infection and proliferation.
The earliest vBcl-2 homologs identified were those discovered by the presence of their characteristic BH sequence motifs and these regions were confirmed to be vital for their function [42]. Viral Bcl-2 members included in this group are those of E1B19K from adenovirus [43][44], the herpesviruses (herpesviridae) Epstein Barr virus (EBV), BHRF1 [45][46], Kaposi sarcoma virus, KsBcl2 [47], turkey herpes virus vNR13 (a viral Bcl-B ortholog) [48], Herpesvirus saimiri ORF16 [49], and murine γ-herpes virus 68/M11 [50]. Bcl-2 genes have also been identified in asfarviridae (African swine fever virus, ASFV A179L) [51][52], and iridiviridae (Grouper Iridovirus GIV66 [53]). Saliently, poxviridae members such as vaccinia virus (VACV) have been shown to bear Bcl-2 genes (VACV F1L) [38][54], however, the lack of identifiable primary sequence identity with cellular Bcl-2 proteins for many of the poxvirus encoded genes hampered their identification as bona fide Bcl-2 proteins.
Poxviruses have relatively large and complex genomes when compared to other viruses, and employ multiple strategies for modulating host-cell apoptosis [7]. Frequently, poxviruses contain multiple Bcl-2 mimics (e.g., VACV N1L and F1L are both Bcl-2 mimics) that interfere with Bax-Bak regulated apoptosis that attests to the importance of manipulating this pathway. Other strategies employed by poxviruses include TNF receptor homologs such as CrmB, CrmC, CrmD, and CrmE [55], Serine protease inhibitors (CPXV CrmA, VACV B13) [56][57], Golgi anti-apoptotic protein GAAP [58], double stranded RNA (dsRNA) induced apoptosis (e.g., VACV E3, MYXV M029, SPV032) [59][60] and Cu–Zn–Superoxide dismutase (SOD) induced apoptosis inhibition (M131, S131) [61]. These strategies are summarized in Figure 2. However, there is significantly less structural and interaction data available for these non-Bcl-2 mimics. Apart from potential health risk from poxviruses, current research is focusing on various immunomodulatory strategies encoded by various poxviruses against host immune systems [7][62]. This review focuses on the role of poxviral Bcl-2 proteins.
2. Pox Virus Inhibition of Host Intrinsic Activated Apoptosis with Bcl-2 Homologs
Poxviridae are a sizeable and diverse group of viruses that infect both vertebrates and invertebrates and are subdivided into the entomopoxviridae, which infect invertebrates such as insects, and chordopoxviridae, which infect vertebrates [63]. Ten genera of poxviridae are currently identified and classified under chordopoxviridae [63]. These are: orthopoxvirus, capripoxvirus, cervidpoxvirus, suipoxvirus, leporipoxvirus, mollusicpoxvirus, yatapoxvirus, avipoxvirus, crocodylidpoxvirus, and parapoxvirus. Among these phyla, orthopoxvirus, molluscipoxvirus, yatapoxvirus, and parapoxvirues have been shown to infect humans and cause disease [64]. For example, monkeypox virus is an orthopoxvirus and is classified as an emerging zoonotic disease that could have a potentially significant impact on human health [65].
Poxviruses are large linear double stranded DNA viruses that contain 135–360 kbp, which encode up to 328 open reading frames (ORFs) [66] and exclusively replicate within the cytoplasm of the infected cells [63]. Perhaps the two most well-known examples of the pox family are variola virus (VARV), the causative agent responsible for smallpox and vaccinia virus (VACV), the vehicle for delivery of the smallpox vaccine [67]. VARV and VACV are closely related orthopoxviruses [68] that bear multiple immunomodulatory genes including Bcl-2 homologs. [38][69][70][71][72]. Almost all poxviridae of the chordopox families encode Bcl-2 like proteins and significantly no entomopox viruses (insect infecting) have yet been identified with Bcl-2 homologs in their genomes. This observation is likely due to differences in Bcl-2 mediated apoptosis in invertebrates compared to vertebrates [1][19]. Here, we review the state of knowledge on the Bcl-2 genes in the chordopox viruses and their structural biology, interactions with the cellular pro-apoptotic Bcl-2 members, and mechanisms of action to block host apoptosis, thus shedding light on how chordopox viruses successfully infect and replicate inside host cells.
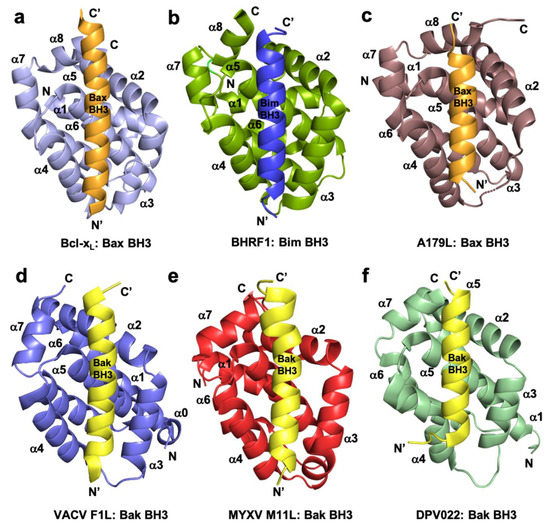
Figure 1. Cartoon representation of crystal structures of poxviral encoded Bcl-2 proteins and comparison with cellular Bcl-xL and other viral Bcl-2 structures. (a) Human Bcl-xL (light blue) in complex with Bax BH3 (orange) [73]. All helices are labelled as α1-α7/8 and the view is into the canonical ligand binding groove formed by helices α2-α5. (b) EBV BHRF1 (split pea) in complex with Bim BH3 (blue) [45], (c) ASFV A179L (dirty violet) in complex with Bax BH3 [52] (orange), (d) VACV F1L (slate) bound to Bak BH3 (yellow) [54], (e) MYXV M11L (red) bound to Bak BH3 (yellow) [37], and (f) DPV022 (wheat) in complex with Bak BH3 (yellow) [74]. The orientations depicted in (b)–(f) are identical to that in (a).
2.1. Orthopoxviruses
VACV, the prototypical member of orthopoxviridiae, bears multiple proteins that have either been established or predicted to have structures similar to the Bcl-2-fold [75]. Examples of structures of this class of protein have been given in Figure 3.
VACV F1L was the first identified Bcl-2 like protein linked to mitochondria associated apoptosis inhibition in vaccinia virus [76], but shares no recognizable sequence identity with any mammalian Bcl-2 family members. Functional studies of VACV F1L revealed that it is an essential element for survival of virus infected cells and prevented staurosporin induced cell death and subsequent cytochrome c release from mitochondria in Jurkat cells [76]. In comparison, an f1l deletion mutant, VV811, underwent apoptosis and expression of F1L prevented all post-mitochondrial events [76][77]. Similar to other pro-survival Bcl-2 proteins, VACV F1L was also shown to localize to mitochondrial membranes through its hydrophobic C-terminal residues [76]. Biochemical interaction studies of VACV F1L revealed that it has a highly selective BH3 binding profile and is only able to bind to peptides that span the BH3 regions from Bim, Bak, and Bax with sub micromolar affinities [38]. Interaction of VACV F1L with Bim is key to its pro-survival activity [54]. Mutagenesis of the F1L binding groove residue, A115W, hindered the interactions with BimL, but not with Bak and the mutant was unable to prevent host cell apoptosis [54]. In addition, F1L prevented Bak and Bax homo-oligomerization and subsequent cell death [77], in part by replacing the activity of Mcl-1 [78]. The crystal structure of F1L featured the conserved Bcl-2 fold with seven α-helices where helices α2–α5 form the canonical ligand binding groove, but the overall fold was a dimer featuring an unusual domain swapped configuration where the α1 helix of two neighboring protomers were swapped (Figure 3). In contrast to F1L, mammalian pro-survival Bcl-2 protein possesses eight α-helices that feature an additional short helix near the C-terminus. In addition to the domain swapped dimer conformation, F1L features a unique N-terminal extension spanning residues 1–56. It was previously reported that this extension harbors caspase-9 inhibitory activity [79] and was predicted to be helical [80]. However, a subsequent study demonstrated that the N-terminal extension was intrinsically disordered and did not contribute to apoptosis regulation [38][81]. More recent functional analysis of this N-terminal region suggested an inflammasome inhibitory function via direct interaction with NLRP1 [82], which is important for initiating innate immune responses against invading pathogens [83] (Figure 2). VARV also encodes an F1L homolog, VARV F1L, and although it has an almost identical structure and sequence to VACV F1L [39], it differs functionally from VACV F1L. VARV F1L inhibits host apoptosis through interacting with only Bid, Bak, and Bax but does not bind Bim (Table 1). Compared to VACV F1L, VARV F1L only inhibits Bax mediated apoptosis but does not inhibit apoptosis via Bak [39].
Other orthopox viruses have not been as well studied and the data available on their vBcl-2 function are much more limited in scope. Ectromelia virus is the causative agent of mousepox and expresses an F1L homolog, EMV025. EMV025 was found to interact with Bak, Bax, and Bim, and blocked the host intrinsic apoptosis pathway by sequestering Bak [84][85].
VACV also bears a second apoptosis inhibitory Bcl-2 like protein in its genome in addition to F1L, the 117-residue protein N1L. N1L has been shown to interact with several cellular pro-apoptotic proteins including Bak, Bid, and Bim with high affinity (Table 1), similar to that observed previously for Bcl-xL [72][86]. In contrast to other vBcl-2 proteins, N1L is localized in the cytosol but not mitochondria and lacks the C-terminal hydrophobic region which in Bcl-2 proteins targets the outer mitochondrial membrane [72]. The crystal structure of VACV N1L revealed that it adopted an overall Bcl-2 fold as a homodimer, where α1 and α6 formed the dimerization interface (Figure 3b) [72]. A structural comparison revealed that VACV F1L and N1L shared a similar structure (Figure 3c). However, functionally VACV N1L inhibits NF-κB signaling during infection as well as block the host intrinsic apoptosis pathway under specific conditions and these functions in N1L are mediated by two different independent sites [75][87]. This has been confirmed in transiently transfected N1L immunoprecipitated with cellular Bax, Bid, and Bad, where Bax was expressed by cellular transfection [86].
Regardless of their structural similarity to Bcl-2 proteins, Bcl-2 homologs do not necessarily involve manipulation of host cell apoptosis; most orthopox Bcl-2 homologs are associated with the regulation of host innate immune responses [8] through antagonizing the Toll Like Receptor (TLR) signaling network [88][89] (Figure 2).
VACV A46, which was initially predicted to be a member of the Bcl-2 family [89], and was subsequently shown to adopt a Bcl-2 fold comprising seven α-helices [90][91]. Notably, A46 does not harbor Bcl-2 like anti-apoptotic activity, instead, A46 is an inhibitor of the Toll/interleukin-1 receptor (TIR)-domain adaptor protein, which is crucial for triggering innate immune responses against invading pathogens [90][92][93]. Thus, VACV A46 particularly targets the TIR in a region known as the BB-loop, which is a well conserved short peptide sequence (30 residues) of TIR-domain proteins [94] that has no shared sequence identity with BH3 motifs and blocks the interactions between receptor and adaptor [91] and downstream activation of NF-κB signaling [90]. The crystal structure of A46 showed that it exists as a homodimer with a Bcl-2 fold similar to that seen in VACV N1L [72], A52 [95], B14 [95], and K7 (Figure 3) [91][96], where the α2 helix of one protomer interacts with the α6 helix of the neighboring protomer to form the dimer interface [91][95][97]. A46 was shown to interact with the TIR motif of adaptor protein MAL (MyD88-adaptor-like protein) with 13 uM affinity via the α1 helix of A46, and mutational analysis showed that the mutant E97A reduced the affinity by around 36-fold and K88A reduced by 4-fold, in which E97 played a significant role [91]. Recent data revealed that A46 interferes with the formation of filaments from TIR domains of both MAL and MyD88, two TLR adaptor proteins whose filaments trigger the early activation of NF-κB. Mutagenesis data mapped the interaction site of A46 with MAL/MyD88 filaments to a region spanning a-helices 1 and 7 as well as the flexible C-terminus, thus providing a mechanistic insight into A46 mediated inhibition of NF-κB activation [98].
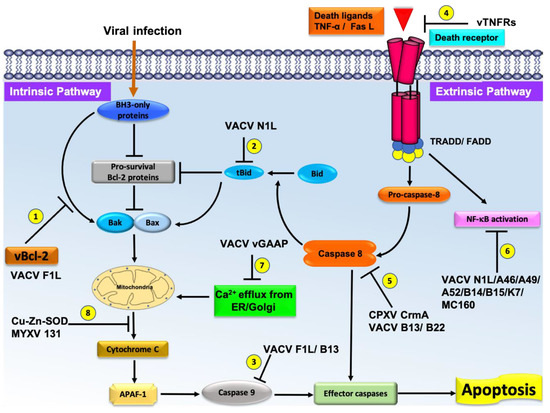
Figure 2. Schematic diagram of the major pathways of apoptosis and the virus encoded effector molecules that modulated them. All major apoptosis inhibition mechanisms utilized by poxviruses are numbered as 1–8. The cellular intrinsic apoptosis pathway is initiated in the event of viral infection, leading to activation of BH3-only proteins in the cytoplasm. These BH3 proteins interact with cellular pro-survival Bcl-2 proteins to neutralize their activity or directly interact with cellular pro-apoptotic Bcl-2 proteins, releasing them to oligomerize at the mitochondrial outer membrane, leading to membrane permeabilization. Release of cytochrome c and other factors from mitochondria activates the downstream events of intrinsic apoptosis such as APAF-1 activation. Virally encoded Bcl-2 proteins mimic the action of cellular pro-survival Bcl-2 proteins to block apoptosis by a variety of mechanisms. (1) VACV F1L interacts with BH3-only proteins to block their activity and subsequent activation of mitochondrial mediated apoptosis. (2) VACV N1L interacts with cellular BH3-only protein Bid to inhibit the downstream activation of apoptosis. (3) VACV F1L/VACV B13 interacts with caspase-9 and suppress the activation of the caspase cascade and subsequent cell death. (4) Poxvirus encoded vTNFRs mimic cellular TNFR1/2 and block the initiation of extrinsic apoptosis. (5) Poxvirus encoded CPXV CrmA/ VACV B13/B22 antagonize active caspase-8 and downstream activation of intrinsic or extrinsic apoptosis. (6) VACV N1L/A46/A49/A52/B14/B15/K7/MC160 inhibit the activation of NF-κB activation, (7) VACV GAAP blocks the Ca2+ efflux from Golgi/ER, and (8) Cu–Zn–SOD encoded by MYXV inhibits the MOMP and subsequent activation of intrinsic apoptosis.
VACV A52 and B14 are intracellular 23 kDa and 17 kDa proteins, respectively, which were predicted to have Bcl-2 folds by secondary structure prediction [89]. VACV A52 and B14 express early in the infection cycle and are important virulence factors that function by inhibiting the activation of NF-κB signaling [95][99] (Figure 2). Structural and solution analysis of VACV A52 and B14 (Figure 3d,e) showed them to homodimerize both in-vitro as well as in the crystal structure as previously reported for VACV N1 and A46 [95]. Neither A52 nor B14 form a hydrophobic binding groove that is required for interaction with the BH3 motif of pro-apoptotic Bcl-2 proteins [95]. An in-vivo transfection analysis reported that both A52 and B14 have a function in NF-κB signaling [95] though there is yet to be any structural analysis. Nevertheless, structure based phylogenetic analysis proposed that A52 and B14 are more similar to VACV N1 rather than Bcl-2 proteins, although they adopt a similar Bcl-2 fold [95].
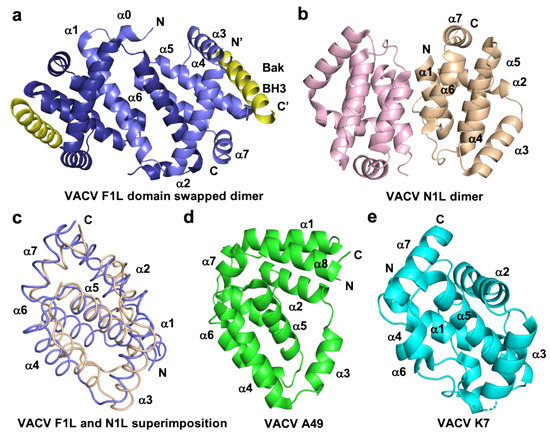
Figure 3. Cartoon representation of different topologies of VACV Bcl-2 homologs. (a) Crystal structure of the dimer formed by VACV F1L (slate) in complex with Bak BH3 (yellow) where α1 helix of one protomer is swapped with α1 helix of a neighboring protomer, (b) dimer interface formed by VACV N1L (beige), where α1 helix of one protomer interact with α6 of the adjacent protomer in crystal contact. (c) Structural superimposition of cartoon tube representation of VACV F1L and VACV N1L as in a view of hydrophobic binding groove made by α2-α5 facing to front. (d) Cartoon representation of A49 (green) and (e) structure of K7 (cyan). Helices are labelled α1–α7 in (a)–(e).
VACV K7 is a 17 kDa intracellular Bcl-2 like protein that does not feature the canonical ligand binding groove unique to Bcl-2 proteins (Figure 3) and has been shown to interact with IRAK2 and TRAF6 in in vivo transfected assays to downregulate the activation of TLR-dependent NF-κB [100]. VACV K7 has been shown to interact with DEAD-box RNA helicase (DDX3), which plays an important role in the innate immune response [101] and binds VACV K7 with an affinity of 510 nM as determined from ITC experiments [101]. The crystal structure of VACV K7 in complex with DDX3 revealed that the DDX3 binding pocket on K7 is located in the region covered by the N-terminus of α1 helix and the α6 helix, and is largely unstructured [101]. These data suggest vaccinia virus has captured a Bcl-2 like gene and over time and adapted it for various immunomodulatory functions.
VACV A49 is a 19 kDa Bcl-2 like protein predominantly expressed in the cytosol and does not possess pro-survival function. A49 also does not feature a hydrophobic ligand binding groove, and structurally resembles MYXV M11L [102] (Figure 3c). However, homologs of A49 are only found in orthopoxviral genomes [103] and have been shown to inhibit NF-κB activation and translocation into the nucleus, with the A49 knockout virus unable to block the NF-κB activation [104]. Similar to VACV K7, A49 exists as a monomer in solution or in cells [104].
VACV encodes another inhibitor of intrinsic apoptosis, M1, which has not been structurally characterized [105], but is predicted to be an ankyrin-repeat like (ANK) protein. Mechanistically, M1L was shown to inhibit staurosporin-induced apoptosis, and co-immunoprecipitated with APAF-1: Caspase-9 complexes [105].
2.2. Leporipoxviruses
Leporipoxviruses (Leporipoxviridae) cause disease in rabbits and squirrels, and comprise four members with myxoma virus (MV) being considered the prototypical member of the genus [64]. MV is the causative agent of myxomatosis in European rabbits [106] and encodes a vBcl-2 like protein, M11L, which is localized to the MOM via its hydrophobic C-terminal region. The crystal structure of M11L showed that it adopts a monomeric Bcl-2 fold despite the lack of detectable primary sequence identity with cellular Bcl-2 proteins [37][107]. The structure of M11L features seven α-helices and a hydrophobic binding groove that engages with pro-apoptotic proteins, similar to that in other pro-survival proteins [37][107]. M11L displays high affinity toward the BH3-only protein Bim and was able to bind other pro-apoptotic proteins Bak [108], Bax, Bid, and Bmf with high to moderate affinity (Table 1) [37]. Cellular studies confirmed that, unlike VACV F1L, M11L subverts host cell apoptosis by primarily sequestering cellular Bak and Bax [37], but not Bim [54]. Myxoma virus infection initiates a rapid response from cellular Bax, which translocates to mitochondria [106]. Interestingly, expression of M11L could inhibit the Fas-ligand induced apoptosis in HEK293 cells and downregulate the subsequent caspase cascade in virus infected cells [108]. These data suggest that MYXV M11L may inhibit host apoptosis through both the intrinsic and extrinsic pathways.
2.3. Yatapoxviruses
Yatapoxviridae are primate specific poxviruses and two identified members, the tanapoxvirus (TANV) and yaba monkey tumor virus (YLDV), cause mild monkeypox like infections in humans [64]. Genome analysis of both TANV and YLDV revealed a putative Bcl-2 like protein 16L (TANV16L) [109] that shared a 98% sequence identity with each other. TANV16L displayed a broad range of interactions toward pro-apoptotic Bcl-2 proteins by binding to almost all BH3 interactors except Bok and Noxa (Table 1), and inhibited cell death induced by cellular Bax and Bax in a yeast model system [110]. Surprisingly, the crystal structures of TANV16L revealed both monomeric and domain swapped dimeric Bcl-2 configurations where two complexes of TANV16L, Bax BH3, and Bim BH3 crystallized as a domain swapped dimeric configuration and TANV16L: Puma BH3 complex crystallized in a monomeric Bcl-2 fold [110]. This was further validated by an analytical ultracentrifugation experiment showing TANV16L exists as mainly monomeric and dimeric forms with a minor component of homotetrameric species [110]. The dimeric topology of TANV16L is similar to that observed previously in vaccinia and variola virus Bcl-2 homologs [38][39], with the monomeric configuration only differing due to the α1 helix being folded back into the same protomer.
2.4. Parapoxviruses
Parapoxviridae are also known as epitheliotropic viruses and cause skin infections in humans. ORF virus is the prototypical member of this genus and commonly infects sheep, goats, and humans [111]. ORFV125 is a predicted anti-apoptotic Bcl-2 like gene encoded by the ORF virus [112]. Similar to other vBcl-2 proteins, ORFV125 lacks obvious Bcl-2 homologs, but antagonizes mitochondria mediated apoptosis and caspase activation in virus infected cells [113]. Immunoprecipitation data of ORFV125 revealed that it only engaged with a selective subset of pro-apoptotic proteins: Bim, Puma, Hrk, Bik, Noxa, and active Bax, but not Bak [114]. In contrast, recently reported affinity measurements of ORFV125 showed that it only binds to cellular Bax, Bak, Puma, and Hrk BH3 motif peptides with moderate sub micromolar affinities (Table 1) [115]. However, no interactions with the universal BH3 interactor Bim were detected. Crystal structures of ORFV125 revealed that it exists as a domain swapped dimer [115], similar to the previously reported VACV F1L [38], VARV F1L [39], DPV022 [74], and TANV 16L [110].
2.5. Capripoxviruses
Capripoxviruses are economically important pathogenic viruses that infect domestic ruminants such as sheep and goats. The difficult to control lumpy skin disease is a common disease of sheep caused by sheeppoxvirus, a prototypical member of capripoxvirus that causes significant economic loss by terminating wool production [116]. Sheeppox virus encoded SPPV14 is well characterized capripoxvirus anti-apoptotic vBcl-2 protein that adopts a monomeric Bcl-2 fold similar to M11L [117], and features a broad range of interactions binding all pro-apoptotic host Bcl-2 proteins [118] except Noxa (Table 1).
Table 1. Summary of poxvirus Bcl-2 homolog binding data. Binding affinities of poxviral Bcl-2 proteins measured by isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR). All pox viral proteins were shown to interact with cellular Bak, Bax, and Bim except VARV F1L, which does not interact with Bim. This suggests that poxviruses primarily target cellular Bak and Bax or Bim inhibition and downregulate the subsequent activation of intrinsic apoptosis.
|
Binding Affinities (nM) |
|||||||||
|
Pro-apoptotic protein |
VACV F1L [38] |
VARV F1L [39] |
M11L [37] |
TANV16L [110] |
SPPV14 [117] |
DPV022 [74] |
FPV039 [119] |
CNP058 [120] |
ORFV125 [115] |
|
Bak |
4300 |
2640 |
50 |
38 |
48 |
6930 |
76 |
508 |
5802 |
|
Bax |
1850 |
960 |
75 |
70 |
26 |
4040 |
76 |
326 |
682 |
|
Bok |
N/A |
N/A |
N/A |
NB |
7580 |
N/A |
NB |
NB |
NB |
|
Bad |
NB |
NB |
>1000 |
219 |
5197 |
NB |
653 |
NB |
NB |
|
Bid |
NB |
3200 |
100 |
719 |
136 |
NB |
2 |
50 |
NB |
|
Bik |
NB |
NB |
>1000 |
1250 |
1766 |
NB |
30 |
NB |
NB |
|
Bim |
250 |
NB |
5 |
180 |
19 |
340 |
10 |
353 |
NB |
|
Bmf |
NB |
NB |
100 |
606 |
44 |
NB |
16 |
294 |
NB |
|
Hrk |
NB |
NB |
>1000 |
3220 |
39 |
NB |
24 |
312 |
1912 |
|
Noxa |
NB |
NB |
>1000 |
NB |
NB |
NB |
28 |
3284 |
NB |
|
Puma |
NB |
NB |
>1000 |
468 |
56 |
NB |
24 |
2484 |
1753 |
NB-No Binding, N/A- Not Available.
Deerpox virus DPV022 is another apoptosis inhibitory Bcl-2 gene identified in capripoxviruses and does not feature obvious BH motifs. DPV022 was shown to block intrinsic apoptosis by interacting with a highly selective subset of pro-apoptotic protein Bak, Bax [121], and Bim. The structure of DPV022 revealed a domain swapped Bcl-2 fold [74], which has previously been seen in vaccinia and variola virus F1L [38][39][54].
2.6. Avipoxviruses
Avipoxviruses are a group of prominent pathogenic viruses among avian species, which cause a slower growth in birds [122]. Among the sequenced genomes of avipoxviridae genus, putative Bcl-2 proteins of two members have been characterized, FPV039 from fowlpox virus (FPV) and CNP058 from canarypox virus (CNPV). Both of these proteins adopt the conserved monomeric Bcl-2 fold with seven alpha helices [119][120]. FPV039 is able to suppress the host apoptotic machinery by engaging with all pro-apoptotic Bcl-2 proteins (Table 1) [119], and the FPV039:Bax interaction prevents Bax oligomerization and mitochondrial pore-formation [123]. In contrast, CNP058 interacts with a distinct set of BH3-only proteins and did not show any detectable affinity toward Bok, Bad, or Bik (Table 1) [120]. Thus, both FPV039 and CNP059 potentially block host apoptosis by sequestering Bim and direct interactions with cellular Bak and Bax [119][120].
3. Conclusions
With their relatively complex genomes, poxviruses have developed a diverse array of anti-apoptotic strategies to block host cell suicide mechanisms. However, though there are many apoptotic regulating genes in the poxviruses genomes there is a paucity of direct interaction measurements or structural data to indicate how they function. Perhaps the exception to the lack of functional and structural data arises from those of the Bcl-2 family that regulate mitochondrial initiated apoptosis. A multitude of biological, biochemical and structural experiments on viral Bcl-2 proteins has shown they engage with crucial host apoptosis signaling proteins. It is now clear that there are multiple modes of apoptosis inhibition by the viral Bcl-2 homologs and perhaps makes this class of viral apoptosis inhibitor the most well understood at a molecular and biochemical level. In addition to the numerous described poxviral encoded proteins used to disarm the plethora of apoptosis-associated host responses to viral infection, a number of poxviral modulators of another critical host defense system exist that counter interferon (IFN)-based responses. While the recent structural and biochemical studies described provide a more detailed understanding of apoptosis inhibition by poxviral Bcl-2 proteins, many gaps remain in our understanding of their modulation of host cell apoptosis.
References
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59.
- Kvansakul, M.; Hinds, M.G. The Bcl-2 family: Structures, interactions and targets for drug discovery. Apoptosis 2015, 20, 136–150.
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390.
- Strasser, A.; Vaux, D.L. Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 2017, 25, 13–20.
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683.
- Bugert, J.J.; Darai, G. Poxvirus Homologues of Cellular Genes. In Molecular Evolution of Viruses—Past and Present: Evolution of Viruses by Acquisition of Cellular RNA and DNA; Becker, Y., Darai, G., Eds.; Springer: Boston, MA, USA, 2000; pp. 111–133.
- Nichols, D.B.; De Martini, W.; Cottrell, J. Poxviruses Utilize Multiple Strategies to Inhibit Apoptosis. Viruses 2017, 9, 215.
- Smith, G.L.; Benfield, C.T.O.; De Motes, C.M.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392.
- Smith, G.L.; Talbot-Cooper, C.; Lu, Y. How Does Vaccinia Virus Interfere With Interferon? Adv. Clin. Chem. 2018, 100, 355–378.
- Lawler, C.; Brady, G. Poxviral Targeting of Interferon Regulatory Factor Activation. Viruses 2020, 12, 1191.
- Garcia-Calvo, M.; Peterson, E.P.; Leiting, B.; Ruel, R.; Nicholson, D.W.; Thornberry, N.A. Inhibition of Human Caspases by Peptide-based and Macromolecular Inhibitors. J. Biol. Chem. 1998, 273, 32608–32613.
- Ashkenazi, A. Targeting the extrinsic apoptotic pathway in cancer: Lessons learned and future directions. J. Clin. Investig. 2015, 125, 487–489.
- Schütze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662.
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430.
- Seyrek, K.; Ivanisenko, N.V.; Richter, M.; Hillert, L.K.; König, C.; Lavrik, I.N. Controlling Cell Death through Post-translational Modifications of DED Proteins. Trends Cell Biol. 2020, 30, 354–369.
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.-L.; Schröter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nat. Cell Biol. 1997, 386, 517–521.
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2010, 18, 538–548.
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Mol. Cell 2008, 30, 689–700.
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128.
- Caria, S.; Hinds, M.G.; Kvansakul, M. Structural insight into an evolutionarily ancient programmed cell death regulator—The crystal structure of marine sponge BHP2 bound to LB-Bak-2. Cell Death Dis. 2018, 8, e2543.
- Popgeorgiev, N.; Sa, J.D.; Jabbour, L.; Banjara, S.; Nguyen, T.T.M.; Akhavan-E-Sabet, A.; Gadet, R.; Ralchev, N.; Manon, S.; Hinds, M.G.; et al. Ancient and conserved functional interplay between Bcl-2 family proteins in the mitochondrial pathway of apoptosis. Sci. Adv. 2020, 6, eabc4149.
- Banjara, S.; Sa, J.D.; Hinds, M.G.; Kvansakul, M. The structural basis of Bcl-2 mediated cell death regulation in hydra. Biochem. J. 2020, 477, 3287–3297.
- Yan, N.; Chai, J.; Lee, E.S.; Gu, L.; Liu, Q.; He, J.; Wu, J.-W.; Kokel, D.; Li, H.; Hao, Q.; et al. Structure of the CED-4–CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nat. Cell Biol. 2005, 437, 831–837.
- Suraweera, C.D.; Caria, S.; Järvå, M.; Hinds, M.G.; Kvansakul, M. A structural investigation of NRZ mediated apoptosis regulation in zebrafish. Cell Death Dis. 2018, 9, 967.
- Vaux, D.L.; Haecker, G.; Strasser, A. An evolutionary perspective on apoptosis. Cell 1994, 76, 777–779.
- Kvansakul, M.; Hinds, M.G. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013, 4, e909.
- Kvansakul, M.; Hinds, M.G. Chapter Three—The Structural Biology of BH3-Only Proteins. In Methods in Enzymology; Ashkenazi, A., Yuan, J., Wells, J.A., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 544, pp. 49–74.
- Luo, X.; O’Neill, K.L.; Huang, K. The third model of Bax/Bak activation: A Bcl-2 family feud finally resolved? F1000Research 2020, 9, 935.
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200.
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 508–520.
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94.
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63.
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092.
- Taylor, J.M.; Barry, M. Near death experiences: Poxvirus regulation of apoptotic death. Virology 2006, 344, 139–150.
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 2002, 3, 1013–1018.
- Hardwick, J.M.; Bellows, D.S. Viral versus cellular BCL-2 proteins. Cell Death Differ. 2003, 10, S68–S76.
- Kvansakul, M.; Van Delft, M.F.; Lee, E.F.; Gulbis, J.M.; Fairlie, W.D.; Huang, D.C.; Colman, P.M. A Structural Viral Mimic of Prosurvival Bcl-2: A Pivotal Role for Sequestering Proapoptotic Bax and Bak. Mol. Cell 2007, 25, 933–942.
- Kvansakul, M.; Yang, H.; Fairlie, W.D.; Czabotar, P.E.; Fischer, S.F.; Perugini, M.A.; Huang, D.C.S.; Colman, P.M. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008, 15, 1564–1571.
- Marshall, B.J.; Puthalakath, H.; Caria, S.; Chugh, S.S.; Doerflinger, M.; Colman, P.M.; Kvansakul, M. Variola virus F1L is a Bcl-2-like protein that unlike its vaccinia virus counterpart inhibits apoptosis independent of Bim. Cell Death Dis. 2015, 6, e1680.
- Altmann, M.; Hammerschmidt, W. Epstein-Barr Virus Provides a New Paradigm: A Requirement for the Immediate Inhibition of Apoptosis. PLoS Biol. 2005, 3, e404.
- Kvansakul, M.; Caria, S.; Hinds, M.G. The Bcl-2 Family in Host-Virus Interactions. Viruses 2017, 9, 290.
- Chiou, S.K.; Tseng, C.C.; Rao, L.; White, E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 1994, 68, 6553–6566.
- White, E.; Cipriani, R.; Sabbatini, P.; Denton, A. Adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J. Virol. 1991, 65, 2968–2978.
- Han, J.; Sabbatini, P.; Perez, D.; Rao, L.; Modha, D.; White, E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996, 10, 461–477.
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.S.; Colman, P.M. Structural Basis for Apoptosis Inhibition by Epstein-Barr Virus BHRF1. PLoS Pathog. 2010, 6, e1001236.
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668.
- Cheng, E.H.-Y.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.-G.; Reitz, M.S.; Hardwick, J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 1997, 94, 690–694.
- Reddy, V.R.A.P.; Sadigh, Y.; Tang, N.; Yao, Y.; Nair, V. Novel Insights into the Roles of Bcl-2 Homolog Nr-13 (vNr-13) Encoded by Herpesvirus of Turkeys in the Virus Replication Cycle, Mitochondrial Networks, and Apoptosis Inhibition. J. Virol. 2020, 94, 2049-19.
- Nava, V.E.; Cheng, E.H.; Veliuona, M.; Zou, S.; Clem, R.J.; Mayer, M.L.; Hardwick, J.M. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J. Virol. 1997, 71, 4118–4122.
- Ku, B.; Woo, J.-S.; Liang, C.; Lee, K.-H.; Hong, H.-S.; Kim, K.-S.; Jung, J.U.; Oh, B.-H. Structural and Biochemical Bases for the Inhibition of Autophagy and Apoptosis by Viral BCL-2 of Murine γ-Herpesvirus 68. PLoS Pathog. 2008, 4, e25.
- Galindo, I.; Hernaez, B.; Díaz-Gil, G.; Escribano, J.M.; Alonso, C. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology 2008, 375, 561–572.
- Banjara, S.; Caria, S.; Dixon, L.K.; Hinds, M.G.; Kvansakul, M. Structural Insight into African Swine Fever Virus A179L-Mediated Inhibition of Apoptosis. J. Virol. 2017, 91, e02228-16.
- Banjara, S.; Mao, J.; Ryan, T.M.; Caria, S.; Kvansakul, M. Grouper iridovirus GIV66 is a Bcl-2 protein that inhibits apoptosis by exclusively sequestering Bim. J. Biol. Chem. 2018, 293, 5464–5477.
- Campbell, S.; Thibault, J.; Mehta, N.; Colman, P.M.; Barry, M.; Kvansakul, M. Structural Insight into BH3 Domain Binding of Vaccinia Virus Antiapoptotic F1L. J. Virol. 2014, 88, 8667–8677.
- Loparev, V.N.; Parsons, J.M.; Knight, J.C.; Panus, J.F.; Ray, C.A.; Buller, R.M.L.; Pickup, D.J.; Esposito, J.J. A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 3786–3791.
- Kettle, S.; Khanna, A.; Alcam, A.; Jassoy, C.; Ehret, R.; Smith, G.L. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J. Gen. Virol. 1997, 78, 677–685.
- Macen, J.L.; Upton, C.; Nation, N.; McFadden, G. SERP1, a Serine Proteinase Inhibitor Encoded by Myxoma Virus, Is a Secreted Glycoprotein That Interferes with Inflammation. Virology 1993, 195, 348–363.
- Saraiva, N.; Prole, D.L.; Carrara, G.; De Motes, C.M.; Johnson, B.; Byrne, B.; Taylor, C.W.; Smith, G.L. Human and Viral Golgi Anti-apoptotic Proteins (GAAPs) Oligomerize via Different Mechanisms and Monomeric GAAP Inhibits Apoptosis and Modulates Calcium. J. Biol. Chem. 2013, 288, 13057–13067.
- Kibler, K.V.; Shors, T.; Perkins, K.B.; Zeman, C.C.; Banaszak, M.P.; Biesterfeldt, J.; Langland, J.O.; Jacobs, B.L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 1997, 71, 1992–2003.
- González-Santamaría, J.; Campagna, M.; Garcia, M.A.; Marcos-Villar, L.; González, D.; Gallego, P.; Lopitz-Otsoa, F.; Guerra, S.; Rodríguez, M.S.; Esteban, M.; et al. Regulation of Vaccinia Virus E3 Protein by Small Ubiquitin-Like Modifier Proteins. J. Virol. 2011, 85, 12890–12900.
- Teoh, M.L.T.; Walasek, P.J.; Evans, D.H. Leporipoxvirus Cu,Zn-Superoxide Dismutase (SOD) Homologs Are Catalytically Inert Decoy Proteins That Bind Copper Chaperone for SOD. J. Biol. Chem. 2003, 278, 33175–33184.
- Weaver, J.R.; Isaacs, S.N. Monkeypox virus and insights into its immunomodulatory proteins. Immunol. Rev. 2008, 225, 96–113.
- Moss, B. Poxvirus DNA Replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010199.
- Barrett, J.W.; McFadden, G. Chapter 19—Origin and Evolution of Poxviruses. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008; pp. 431–446.
- Mccollum, A.M.; Damon, I.K. Human Monkeypox. Clin. Infect. Dis. 2013, 58, 260–267.
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40.
- Fenner, F. Risks and benefits of vaccinia vaccine use in the worldwide smallpox eradication campaign. Res. Virol. 1989, 140, 465–466.
- Reynolds, M.G.; Guagliardo, S.A.; Nakazawa, Y.J.; Doty, J.B.; Mauldin, M.R. Understanding orthopoxvirus host range and evolution: From the enigmatic to the usual suspects. Curr. Opin. Virol. 2018, 28, 108–115.
- Barry, M.; Wasilenko, S.T.; Stewart, T.L.; Taylor, J.M. Apoptosis Regulator Genes Encoded by Poxviruses. In Viruses and Apoptosis. Progress in Molecular and Subcellular Biology; Springer: Berlin/Heidelberg, Germany, 2004; Volume 36, pp. 19–37.
- Everett, H.; McFadden, G. Poxviruses and apoptosis: A time to die. Curr. Opin. Microbiol. 2002, 5, 395–402.
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses Andimmuneevasion. Annu. Rev. Immunol. 2003, 21, 377–423.
- Aoyagi, M.; Zhai, D.; Jin, C.; Aleshin, A.E.; Stec, B.; Reed, J.C.; Liddington, R.C. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007, 16, 118–124.
- Czabotar, P.E.; Lee, E.F.; Thompson, G.V.; Wardak, A.Z.; Fairlie, W.D.; Colman, P.M. Mutation to Bax beyond the BH3 Domain Disrupts Interactions with Pro-survival Proteins and Promotes Apoptosis. J. Biol. Chem. 2011, 286, 7123–7131.
- Burton, D.R.; Caria, S.; Marshall, B.; Barry, M.; Kvansakul, M. Structural basis of Deerpox virus -mediated inhibition of apoptosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1593–1603.
- Veyer, D.L.; Carrara, G.; De Motes, C.M.; Smith, G.L. Vaccinia virus evasion of regulated cell death. Immunol. Lett. 2017, 186, 68–80.
- Wasilenko, S.T.; Stewart, T.L.; Meyers, A.F.A.; Barry, M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 14345–14350.
- Fischer, S.F.; Ludwig, H.; Holzapfel, J.; Kvansakul, M.; Chen, L.; Huang, D.C.S.; Sutter, G.; Knese, M.; Häcker, G. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain-binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ. 2005, 13, 109–118.
- Campbell, S.; Hazes, B.; Kvansakul, M.; Colman, P.; Barry, M. Vaccinia Virus F1L Interacts with Bak Using Highly Divergent Bcl-2 Homology Domains and Replaces the Function of Mcl-1. J. Biol. Chem. 2009, 285, 4695–4708.
- Zhai, D.; Yu, E.; Jin, C.; Welsh, K.; Shiau, C.-W.; Chen, L.; Salvesen, G.S.; Liddington, R.; Reed, J.C. Vaccinia Virus Protein F1L Is a Caspase-9 Inhibitor. J. Biol. Chem. 2009, 285, 5569–5580.
- Yu, E.; Zhai, D.; Jin, C.; Gerlic, M.; Reed, J.C.; Liddington, R. Structural Determinants of Caspase-9 Inhibition by the Vaccinia Virus Protein, F1L. J. Biol. Chem. 2011, 286, 30748–30758.
- Caria, S.; Marshall, B.; Burton, R.-L.; Campbell, S.; Pantaki-Eimany, D.; Hawkins, C.J.; Barry, M.; Kvansakul, M. The N Terminus of the Vaccinia Virus Protein F1L Is an Intrinsically Unstructured Region That Is Not Involved in Apoptosis Regulation. J. Biol. Chem. 2016, 291, 14600–14608.
- Gerlic, M.; Faustin, B.; Postigo, A.; Yu, E.C.-W.; Proell, M.; Gombosuren, N.; Krajewska, M.; Flynn, R.; Croft, M.; Way, M.; et al. Vaccinia virus F1L protein promotes virulence by inhibiting inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 7808–7813.
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265.
- Mehta, N.; Taylor, J.; Quilty, D.; Barry, M. Ectromelia virus encodes an anti-apoptotic protein that regulates cell death. Virology 2015, 475, 74–87.
- Antignani, A.; Youle, R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689.
- Cooray, S.; Bahar, M.W.; Abrescia, N.G.A.; McVey, C.E.; Bartlett, N.W.; Chen, R.A.-J.; Stuart, D.I.; Grimes, J.M.; Smith, G.L. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007, 88, 1656–1666.
- De Motes, C.M.; Cooray, S.; Ren, H.; Almeida, G.M.F.; McGourty, K.; Bahar, M.W.; Stuart, D.I.; Grimes, J.M.; Graham, S.C.; Smith, G.L. Inhibition of Apoptosis and NF-κB Activation by Vaccinia Protein N1 Occur via Distinct Binding Surfaces and Make Different Contributions to Virulence. PLoS Pathog. 2011, 7, e1002430.
- DiPerna, G.; Stack, J.; Bowie, A.G.; Boyd, A.; Kotwal, G.; Zhang, Z.; Arvikar, S.; Latz, E.; Fitzgerald, K.A.; Marshall, W.L. Poxvirus Protein N1L Targets the I-κB Kinase Complex, Inhibits Signaling to NF-κB by the Tumor Necrosis Factor Superfamily of Receptors, and Inhibits NF-κB and IRF3 Signaling by Toll-like Receptors. J. Biol. Chem. 2004, 279, 36570–36578.
- González, J.M.; Esteban, M. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: Sequence similarity and evolutionary history. Virol. J. 2010, 7, 59.
- Fedosyuk, S.; Grishkovskaya, I.; Ribeiro, E.D.A.; Skern, T. Characterization and Structure of the Vaccinia Virus NF-κB Antagonist A46. J. Biol. Chem. 2013, 289, 3749–3762.
- Kim, Y.; Lee, H.; Heo, L.; Seok, C.; Choe, J. Structure of vaccinia virus A46, an inhibitor of TLR4 signaling pathway, shows the conformation of VIPER motif. Protein Sci. 2014, 23, 906–914.
- O’Neill, L.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364.
- Fedosyuk, S.; Bezerra, G.A.; Radakovics, K.; Smith, T.K.; Sammito, M.; Bobik, N.; Round, A.; Eyck, L.F.T.; Djinović-Carugo, K.; Usón, I.; et al. Vaccinia Virus Immunomodulator A46: A Lipid and Protein-Binding Scaffold for Sequestering Host TIR-Domain Proteins. PLoS Pathog. 2016, 12, e1006079.
- Toshchakov, V.Y.; Fenton, M.J.; Vogel, S.N. Cutting Edge: Differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J. Immunol. 2007, 178, 2655–2660.
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.-J.; Whalen, D.M.; Abrescia, N.G.A.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia Virus Proteins A52 and B14 Share a Bcl-2–Like Fold but Have Evolved to Inhibit NF-κB rather than Apoptosis. PLoS Pathog. 2008, 4, e1000128.
- Kalverda, A.P.; Thompson, G.S.; Vogel, A.; Schröder, M.; Bowie, A.G.; Khan, A.R.; Homans, S.W. Poxvirus K7 Protein Adopts a Bcl-2 Fold: Biochemical Mapping of Its Interactions with Human DEAD Box RNA Helicase DDX3. J. Mol. Biol. 2009, 385, 843–853.
- Tang, Q.; Chakraborty, S.; Xu, G. Mechanism of vaccinia viral protein B14–mediated inhibition of IκB kinase β activation. J. Biol. Chem. 2018, 293, 10344–10352.
- Azar, D.F.; Haas, M.; Fedosyuk, S.; Rahaman, H.; Hedger, A.; Kobe, B.; Skern, T. Vaccinia Virus Immunomodulator A46: Destructive Interactions with MAL and MyD88 Shown by Negative-Stain Electron Microscopy. Structure 2020, 28, 1271–1287.e5.
- Harte, M.T.; Haga, I.R.; Maloney, G.; Gray, P.; Reading, P.C.; Bartlett, N.W.; Smith, G.L.; Bowie, A.G.; O’Neill, L.A.J. The Poxvirus Protein A52R Targets Toll-like Receptor Signaling Complexes to Suppress Host Defense. J. Exp. Med. 2003, 197, 343–351.
- Schröder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKε-mediated IRF activation. EMBO J. 2008, 27, 2147–2157.
- Oda, S.-I.; Schröder, M.; Khan, A.R. Structural Basis for Targeting of Human RNA Helicase DDX3 by Poxvirus Protein K7. Structure 2009, 17, 1528–1537.
- Neidel, S.; Ren, H.; Torres, A.A.; Smith, G.L. NF-κB activation is a turn on for vaccinia virus phosphoprotein A49 to turn off NF-κB activation. Proc. Natl. Acad. Sci. USA 2019, 116, 5699–5704.
- Mansur, D.S.; De Motes, C.M.; Unterholzner, L.; Sumner, R.P.; Ferguson, B.J.; Ren, H.; Strnadova, P.; Bowie, A.G.; Smith, G.L. Poxvirus Targeting of E3 Ligase β-TrCP by Molecular Mimicry: A Mechanism to Inhibit NF-κB Activation and Promote Immune Evasion and Virulence. PLoS Pathog. 2013, 9, e1003183.
- Neidel, S.; De Motes, C.M.; Mansur, D.S.; Strnadova, P.; Smith, G.L.; Graham, S.C. Vaccinia Virus Protein A49 Is an Unexpected Member of the B-cell Lymphoma (Bcl)-2 Protein Family. J. Biol. Chem. 2015, 290, 5991–6002.
- Ryerson, M.R.; Richards, M.M.; Kvansakul, M.; Hawkins, C.J.; Shisler, J.L. Vaccinia Virus Encodes a Novel Inhibitor of Apoptosis That Associates with the Apoptosome. J. Virol. 2017, 91, e01385-17.
- Su, J.; Wang, G.; Barrett, J.W.; Irvine, T.S.; Gao, X.; McFadden, G. Myxoma Virus M11L Blocks Apoptosis through Inhibition of Conformational Activation of Bax at the Mitochondria. J. Virol. 2006, 80, 1140–1151.
- Douglas, A.E.; Corbett, K.D.; Berger, J.M.; McFadden, G.; Handel, T.M. Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 2007, 16, 695–703.
- Wang, G.; Barrett, J.W.; Nazarian, S.H.; Everett, H.; Gao, X.; Bleackley, C.; Colwill, K.; Moran, M.F.; McFadden, G. Myxoma Virus M11L Prevents Apoptosis through Constitutive Interaction with Bak. J. Virol. 2004, 78, 7097–7111.
- Nazarian, S.H.; Barrett, J.W.; Frace, A.M.; Olsen-Rasmussen, M.; Khristova, M.; Shaban, M.; Neering, S.; Li, Y.; Damon, I.K.; Esposito, J.J.; et al. Comparative genetic analysis of genomic DNA sequences of two human isolates of Tanapox virus. Virus Res. 2007, 129, 11–25.
- Suraweera, C.D.; Anasir, M.I.; Chugh, S.; Javorsky, A.; Impey, R.E.; Zadeh, M.H.; Da Costa, T.P.S.; Hinds, M.G.; Kvansakul, M. Structural insight into tanapoxvirus-mediated inhibition of apoptosis. FEBS J. 2020, 287, 3733–3750.
- Haig, D.; Mercer, A.A. Parapoxviruses. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., Van Regenmortel, M.H.V., Eds.; Academic Press: Oxford, UK, 2008; pp. 57–63.
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; De La Concha-Bermejillo, A.; Lehmkuhl, H.D.; Piccone, M.E.; Kutish, G.F.; Rock, D.L. Genomes of the Parapoxviruses Orf Virus and Bovine Papular Stomatitis Virus. J. Virol. 2004, 78, 168–177.
- Westphal, D.; Ledgerwood, E.C.; Hibma, M.H.; Fleming, S.B.; Whelan, E.M.; Mercer, A.A. A Novel Bcl-2-Like Inhibitor of Apoptosis Is Encoded by the Parapoxvirus Orf Virus. J. Virol. 2007, 81, 7178–7188.
- Westphal, D.; Ledgerwood, E.C.; Tyndall, J.D.A.; Hibma, M.H.; Ueda, N.; Fleming, S.B.; Mercer, A.A. The orf virus inhibitor of apoptosis functions in a Bcl-2-like manner, binding and neutralizing a set of BH3-only proteins and active Bax. Apoptosis 2009, 14, 1317–1330.
- Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. Crystal structures of ORFV125 provide insight into orf virus-mediated inhibition of apoptosis. Biochem. J. 2020, 477, 4527–4541.
- Tuppurainen, E.S.M.; Venter, E.H.; Shisler, J.L.; Gari, G.; Mekonnen, G.A.; Juleff, N.; Lyons, N.A.; De Clercq, K.; Upton, C.; Bowden, T.R.; et al. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound. Emerg. Dis. 2017, 64, 729–745.
- Suraweera, C.D.; Burton, D.R.; Hinds, M.G.; Kvansakul, M. Crystal structures of the sheeppox virus encoded inhibitor of apoptosis SPPV14 bound to the proapoptotic BH3 peptides Hrk and Bax. FEBS Lett. 2020, 594, 2016–2026.
- Okamoto, T.; Campbell, S.; Mehta, N.; Thibault, J.; Colman, P.M.; Barry, M.; Huang, D.C.S.; Kvansakul, M. Sheeppox Virus SPPV14 Encodes a Bcl-2-Like Cell Death Inhibitor That Counters a Distinct Set of Mammalian Proapoptotic Proteins. J. Virol. 2012, 86, 11501–11511.
- Anasir, M.I.; Caria, S.; Skinner, M.A.; Kvansakul, M. Structural basis of apoptosis inhibition by the fowlpox virus protein FPV039. J. Biol. Chem. 2017, 292, 9010–9021.
- Anasir, M.I.; Baxter, A.A.; Poon, I.K.H.; Hulett, M.D.; Kvansakul, M. Structural and Functional Insight into Canarypox Virus CNP058 Mediated Regulation of Apoptosis. Viruses 2017, 9, 305.
- Banadyga, L.; Lam, S.-C.; Okamoto, T.; Kvansakul, M.; Huang, D.C.S.; Barry, M. Deerpox Virus Encodes an Inhibitor of Apoptosis That Regulates Bak and Bax. J. Virol. 2010, 85, 1922–1934.
- Khan, J.S.; Provencher, J.F.; Forbes, M.R.; Mallory, M.L.; Lebarbenchon, C.; McCoy, K.D. Chapter One—Parasites of Seabirds: A Survey of Effects and Ecological Implications. In Advances in Marine Biology; Sheppard, C., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 82, pp. 1–50.
- Banadyga, L.; Veugelers, K.; Campbell, S.; Barry, M. The Fowlpox Virus BCL-2 Homologue, FPV039, Interacts with Activated Bax and a Discrete Subset of BH3-Only Proteins To Inhibit Apoptosis. J. Virol. 2009, 83, 7085–7098.

