 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Johanna Arroyave-Ospina | + 4000 word(s) | 4000 | 2021-02-04 05:01:56 | | | |
| 2 | Karina Chen | Meta information modification | 4000 | 2021-02-18 07:59:37 | | |
Video Upload Options
Oxidative stress (OxS) is considered a major factor in the pathophysiology of inflam-matory chronic liver diseases, including non-alcoholic liver disease (NAFLD). Chronic impairment of lipid metabolism is closely related to alterations of the oxidant/antioxidant balance, which affect metabolism-related organelles, leading to cellular lipotoxicity, lipid peroxidation, chronic endo-plasmic reticulum (ER) stress, and mitochondrial dysfunction. Increased OxS also triggers hepatocytes stress pathways, leading to inflammation and fibrogenesis, contributing to the pro-gression of non-alcoholic steatohepatitis (NASH). The antioxidant response, regulated by the Nrf2/ARE pathway, is a key component in this process and counteracts oxidative stress-induced damage, contributing to the restoration of normal lipid metabolism.
1. Introduction
The maintenance of a “healthy” antioxidant status is essential for cellular homeostasis. Oxidative stress (OxS) is defined as the condition under which the generation of reactive oxygen species (ROS) exceeds the capacity of antioxidants to detoxify. The pathogenesis of several chronic diseases is related to OxS. The liver is an organ where many oxidative processes occur and is, therefore, an important target of OxS-induced damage. Oxidative stress leads to cellular dysfunction, injury, and ultimately cell death, and the impairment of the antioxidant status in the liver contributes significantly to the pathogenesis and progression of chronic liver diseases, including non-alcoholic liver disease (NAFLD) [1]. Various antioxidants have been proposed as a therapeutic agent in liver diseases, in particular non-alcoholic fatty liver disease (NAFLD), based on the clinical and experimental evidence that supports an important role of OxS in the pathophysiology of NAFLD [2].
NAFLD is a pathological condition characterized by fat accumulation in the liver (in more than 5% of hepatocytes) in the absence of alcohol consumption, viral infection, or drugs that can induce steatosis. NAFLD covers a wide spectrum of liver diseases ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), liver fibrosis, and ultimately cirrhosis and hepatocellular carcinoma.
Currently, NAFLD is considered the most prevalent chronic liver disease around the world with a global prevalence of 25% in the general population and an incidence of 28-86/1000 person years. Obesity is among the main risk factors for NAFLD, and NAFLD is often associated with insulin resistance, diabetes mellitus type 2, or metabolic syndrome. NAFLD prevalence is higher than 50% among patients with diabetes type 2 [3]. The accumulation of fat leads to metabolic disturbances, resulting in excessive mitochondrial ROS production and ER stress, and this, in turn, can cause inflammation, cell injury, and cell death. This mechanism is underscored by the fact that NAFLD patients often have an impaired antioxidant status, with decreased serum levels of the antioxidants vitamin E (tocopherol) and vitamin C and increased levels of lipid peroxidation products and systemic oxidative stress markers[4][5].
2. Antioxidant Balance in the Liver in Non-Alcoholic Fatty Liver Disease
2.1. Oxidative Stress Mechanisms in Non-Alcoholic Fatty Liver Disease
Oxidative stress occurs when the balance between oxidants and antioxidants is disrupted. Oxidants or reactive oxygen species (ROS) can be classified as free radicals, which include molecules with one or more unpaired electrons or as non-radical species, generated from two free radical molecules sharing their unpaired electrons. Superoxide anions (O2–), hydroxyl radicals (•OH), and hydrogen peroxide (H2O2) are major physiologically relevant ROS. Superoxide anions are generated by multiple cellular processes, and their production leads to the generation of other oxidant molecules. Superoxide dismutases (SODs) reduce O2– to H2O2, which in turn is converted into hydroxyl radicals (•OH) and water via the iron-catalysed Fenton reaction. Hydroxyl radicals directly or indirectly induce the formation of additional toxic pro-oxidants, including hypochlorous acid, peroxynitrite, and peroxyl radicals [6][7].
ROS are highly toxic molecules and must be detoxified by the antioxidant system. Antioxidant protection systems consist of enzymatic and non-enzymatic components. The enzymatic system includes different enzymes that detoxify ROS. Some of the most relevant are superoxide dismutases (SODs), catalase (CAT), and glutathione peroxidase and reductase (GSH-Px). Non-enzymatic components of the antioxidant system include small molecules such as glutathione, ascorbic acid (vitamin C), retinol (vitamin A), and tocopherol (vitamin E), which act as electron receptors and protect biomolecules and cell structures against damage from ROS. Other important antioxidants include heme oxygenase-1 (HO-1) and redox proteins [1][6].
In NAFLD, there are many sources of OxS. Oxidative phosphorylation (OxPhos) in mitochondria generates ATP, and superoxide anions (O2–) are generated as a by-product of OxPhos. Therefore, OxPhos is an important source of OxS. Increased β-oxidation inside mitochondria and peroxisomes is an additional source of ROS.
In addition to mitochondria, the endoplasmic reticulum (ER) is a source of ROS due to cytochrome P450 (CYP) activity and/or microsomal metabolism or via increased expression of CHOP. It is important to mention that ROS are functional molecules that can also modulate cell signalling and the cellular response to stress [8]. Finally, the inflammatory response also contributes to OxS [9]. During liver injury, OxS induces the activation of redox-sensitive transcription factors, such as NF-kB, Egr-1, and AP-1, leading to an inflammatory response and the activation of cell death pathways in hepatocytes [10][11].
An important component of the antioxidant system is OxS-induced transcription. The nuclear factor E2-related factor 2 (Nrf2) is a redox-sensitive transcription factor and the major regulator of the redox balance. In normal conditions, it is present in the cytoplasm bound to the cytoskeletal-anchoring protein Kelch-like ECH-associated protein 1. High levels of ROS lead to the release of Nrf2 and its translocation to the nucleus to promote the transcription of antioxidant genes, which are regulated by antioxidant response elements (ARE) [12]. Among others, Nrf2 regulates glutathione levels and maintains the reduced glutathione/oxidized glutathione ratio (GSH/GSSG). Moreover, Nrf2 controls the expression of many detoxifying enzymes that eliminate molecules such as H2O2 and peroxide radicals from the cytosol, mitochondria, and the ER [13]. Interestingly, Nrf2 expression appears to be modulated by a variety of inducers (both endogenous as well as exogenous), such as electrophilic agents, redox-active compounds, and xenobiotics [14][15]. Genes that are positively regulated by the Nrf2 pathway include detoxifying enzymes (Phase I, II, and III), redox proteins involved in GSH-based antioxidant mechanisms, and genes related to lipid metabolism [13]
Nrf2 is also involved in lipid metabolism and may play a role in the protection against liver damage during the development of steatosis and steatohepatitis [16]. Nrf2 is important for mitochondrial homeostasis, and it has been demonstrated that Nrf2 activation is necessary to maintain mitochondrial integrity [17] and can directly affect the efficiency of mitochondrial fatty acid oxidation [18]. With regard to lipid accumulation, it has been suggested that Nrf2 represses the expression of key enzymes involved in fatty acid synthesis, thus alleviating hepatic steatosis [19]. Additionally, some in vivo studies using high fat diet (HFD) models of NAFLD showed a negative correlation between Nrf2-induced transcription and hepatic lipogenesis, suggesting that Nrf2 may decrease fatty acid synthesis and lipid accumulation [20][21]. On the other hand, there are also controversial studies that failed to detect an effect on lipid metabolism and even reported that Nrf2 activity increases lipid accumulation [22][23].
The role of Nrf2 in NAFLD is complex and not fully understood yet. Nevertheless, Nrf2 has been proposed as a potential therapeutic target in NAFLD. Nrf2-deficient mice (Nrf2−/− KO) challenged with methionine- and choline-deficient (MCD) diet show exacerbation of liver inflammation and steatosis compared to control mice (Nrf2+/+) [24]. Additionally, Nrf2−/− KO mice, before the start of the MCD diet, showed an altered lipid metabolism, an increased expression of cytochrome P450 enzymes, and lower levels of glutathione, but normal glucose metabolism. Interestingly, in this model, the MCD diet induced an antioxidant response in normal mice but significant oxidative stress in Nrf2−/− KO mice, confirming that OxS, together with impairment of lipid metabolism, is sufficient to drive NAFLD progression even in the absence of insulin resistance. Likewise, restoration of the Nrf2 pathway in Nrf2−/− KO mice improves the fatty liver phenotype [25] mainly by increasing the hepatic antioxidant response and by modulating the expression of lipid-metabolism-related genes such as PPAR α and SREBP1c.
The Nrf2 pathway might be involved in the protection of the liver against OxS and in the pathophysiology of NAFLD. Furthermore, the role of the Nrf2 pathway in lipid accumulation seems to be highly context-dependent and needs further elucidation. Taking into account the importance of the triglyceride/free fatty acid (TG/FFA) balance in NAFLD and the potential role of Nrf2 in modulating this balance, the Nrf2 antioxidant response might be related to an increase in TG synthesis as a protective mechanism. The role of Nrf2 in NAFLD patients is not completely elucidated yet, and its role in disease progression and its potential as a therapeutic target remain unclear. Therefore, studies to evaluate Nrf2 activating compounds for the prevention and treatment of NAFLD are important, and we will discuss some of these novel compounds that are currently being investigated as potential therapies in NAFLD.
The NF-kB pathway has been proposed as a therapeutic target in chronic liver diseases due to its role in OxS-mediated responses [26]. Activation of the NF-kB pathway can mediate protective mechanisms in conditions of OxS, e.g., via reducing ROS generation and the induction of autophagy [27]. Additionally, NF-kB promotes the expression of antioxidant genes such as MnSOD, Glutathione S transferase, and NADPH dehydrogenase [28]. On the other hand, it has been demonstrated that NF-kB signalling is related to both pro-oxidant as well as antioxidant effects and that it is related to the activation of the ER stress response [29]. NF-kB is a redox-sensitive transcription factor, and ROS may modulate its activation through the induction of the pro-inflammatory cytokine TNF-α, while antioxidants such as NAC (N-acetyl-l-cysteine) prevent NF-kB activation [30].
Furthermore, an interaction between NF-kB and the Nrf2 pathway also exists. In general, it has been reported that NF-kB negatively modulates Nrf2 transcription by competition with CBP (CREB-binding protein)–p300 complex [31], and Nrf2 deficiency (knockout) is associated with increased NF-kB activation [32]. Likewise, it has been reported that Nrf2 activation, either in vitro or in vivo, by different antioxidants, prevents inflammation via inhibition of the NF-kB pathway [33][34].
In the context of NAFLD, it has been demonstrated that the loss of Nrf2 leads to hepatic insulin resistance via an NF-kB dependent mechanism [35], showing the pivotal role of OxS and NF-kB-mediated inflammation in the onset of insulin resistance. Likewise, other studies have shown that activation of the Nrf2 pathway, with consequent NF-kB inhibition, improves insulin sensitivity in HFD-fed rats [36].
Modulation of NF-kB signalling by antioxidants has been suggested as a potential therapeutic target in NAFLD, also due to its anti-inflammatory properties. Recent reports show that antioxidant treatment in MCD-induced NASH mice leads to the induction of Nrf2 target genes and the suppression of the NF-kB signalling pathway, resulting in the amelioration of hepatic steatosis, fibrosis, and inflammation [37]. NF-kB modulation independent of Nrf2 by antioxidants such as green tea catechins has also been reported to improve NAFLD [38] by decreasing gut-derived LPS, which decreases inflammation in the liver.
These findings suggest crosstalk between NF-kB and Nrf2 as a promising therapeutic target for NAFLD via inhibition of OxS related signalling.
2.2. Oxidative Stress Biomarkers in NAFLD Patients
Several studies have reported an impaired redox status in the majority of NAFLD patients, as indicated by increased levels of OxS markers and lipid peroxidation products in serum/plasma.
In this regard, other oxidative stress markers have been studied in serum/plasma and liver samples from NAFLD patients, and increased levels/activity for most OxS markers have been reported, such as 8-isoprostane, 8-OH-dG, and TBARS/MDA. NAFLD patients also show increased levels of peroxidised lipids, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which are often used as biomarkers of lipid peroxidation in clinical practice [9]. Moreover, a recent report suggests that reduced levels of free thiol plasma levels can be considered as a global marker of the systemic load of reactive species and can be used as a biomarker for NAFLD [39].
Although most studies show decreased levels of hepatic antioxidant enzymes in NAFLD patients [2], some studies reported conflicting results, showing both increased as well as decreased serum levels of antioxidant enzymes such as SOD, GPx, and GSH in NAFLD patients [40][41]. These conflicting observations could be explained by the pathophysiology of NAFLD in which an initially adaptive antioxidant response to excessive ROS production is followed by exhaustion of the antioxidant system, resulting in lower levels of antioxidant enzymes. Furthermore, a cross-sectional study showed that a high proportion of NAFLD patients had low levels of dietary antioxidants such as vitamin C and retinol, and lower intakes of vitamin A and vitamin E as well [42].
Additional studies comparing the redox status in serum/plasma versus liver tissue at different stages of NAFLD/NASH will be informative to clarify the role of antioxidant components as biomarkers of progression of NAFLD/NASH and their potential as therapeutic targets.
3. Lipotoxicity and Oxidative Stress
There are many mechanisms involved in the development and progression of NAFLD. A schematic overview of these mechanisms is depicted in Figure 1. Excess free fatty acids (FFAs) induce lipid accumulation leading to steatosis and a general impairment of lipid metabolism. In addition, insulin resistance and diabetes type 2 can develop and aggravate the consequences of excess lipid accumulation.
In NAFLD, there is an imbalance in the distribution of different types of lipids between liver cell types, resulting in inflammation triggered by non-parenchymal cells (mainly Kupffer cells), which ultimately leads to hepatocellular damage and fibrogenesis. Indeed, we have previously shown that extracellular vesicles released from steatotic hepatocytes induce an inflammatory response in Kupffer cells and a fibrogenic response in hepatic stellate cells [43]. Direct effects of free fatty acids on Kupffer cells include M1 polarization leading to a pro-inflammatory phenotype [44][45]. Lipotoxicity, defined as a harmful effect of lipid accumulation in non-adipose tissue, is considered a central mechanism in NAFLD progression [46]. The main targets of this lipotoxicity are ER; mitochondria; and signalling pathways, such as JNK and NF-κB, related to inflammation and cell death [47]. Lipotoxicity is to a large extent mediated by OxS phenomena in the liver and will be discussed in the subsequent sections.
Within hepatocytes, FFAs such as palmitate induce lipotoxicity either directly or by increasing the levels of deleterious lipid species such as ceramides and diacylglycerols (DAGs) [48]. Ceramides impair fatty acid oxidation and induce mitochondrial dysfunction [49]. DAGs can activate NF-κB and/or protein kinase C (PKC). Increased lipid peroxidation, impairment of β-oxidation in mitochondria, and the induction of an ER stress response also aggravate the direct toxic effects of lipids [50][51]. The role of ER stress and the Unfolded Protein Response (UPR) in NAFLD is discussed in more detail in Section 3.3.2. The dysfunction of mitochondria and the ER stress induced by impaired lipid metabolism contributes to the generation of ROS, thus causing OxS. The antioxidant balance is further compromised by downregulation of the antioxidant response (e.g., the Nrf2 pathway) and by decreased levels of antioxidant enzymes and molecules (e.g., GSH and SOD).
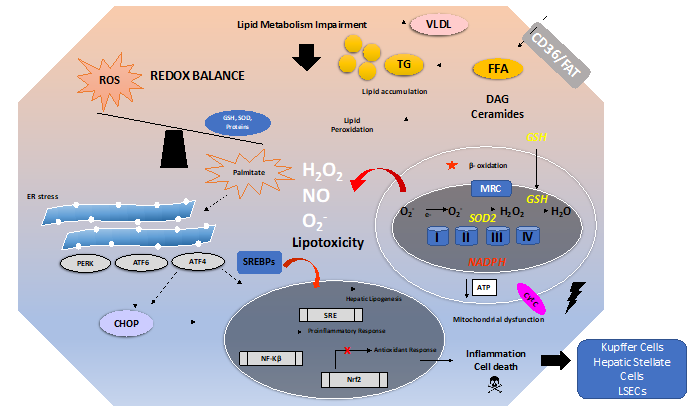
Figure 1. Overview of oxidative stress and antioxidants in the context of non-alcoholic fatty liver disease (NAFLD). The pathogenesis of NAFLD is a multifactorial process involving several mechanisms, ultimately leading to a disturbed redox balance. Impairment of lipid metabolism, e.g., by excessive dietary intake of fat and carbohydrates, leads to steatosis. This can be aggravated by insulin resistance. Free fatty acids (imported by, e.g., CD36 or FAT) such as palmitate cause lipotoxicity by increasing the levels of toxic lipid species such as ceramides and diacylglycerols (DAG). Mitochondrial dysfunction, impairment of β-oxidation, and endoplasmic reticulum (ER) stress can all increase the generation of ROS leading to lipid peroxidation. ER stress induces the UPR, i.e., Unfolded Protein Response. Sustained ER stress and sustained activation of the UPR will trigger activation of the ER stress proteins PERK, ATF6, ATF4, and CHOP, leading to a proinflammatory response and activation of cell death pathways in hepatocytes. ER stress might lead to the activation of sterol regulatory element-binding protein 1C (SREBP1c) and further translocation to the nucleus, thus promoting hepatic lipogenesis. The impaired redox balance also affects the antioxidant response (e.g., the Nrf2 pathway) and leads to decreased levels of antioxidants (e.g., GSH and SOD).
3.1. Lipid Metabolism and Oxidative Stress
In physiological conditions, lipid homeostasis in the liver is maintained by fatty acid uptake (dietary source) and synthesis of triglycerides (TG) versus the catabolic processes involving oxidation and secretion of lipids. Non-esterified fatty acids (NEFAs) can be used as a source of energy, but they can also be esterified into triglycerides either for lipid storage or VLDL synthesis and secretion into the blood. Fatty acid synthesis or de novo lipogenesis occurs in the cytosol, involving several enzymatic pathways. Fatty acid oxidation takes place mainly in mitochondria and to a lesser extent in peroxisomes and in the endoplasmic reticulum (microsomes) [52].
Indeed, it has been demonstrated that hepatic oxidative stress and inflammation are associated with an elevated oxidative metabolism of saturated fatty acids (SFAs) in NAFLD [53]. Some saturated long-chain fatty acids are first oxidized in the peroxisomes and then in the mitochondria. However, these long-chain SFAs are often incompletely oxidized in the mitochondria, affecting metabolic activity and leading to ROS overproduction with subsequent mitochondrial dysfunction [2].
Fatty acid oxidation depends on internal mitochondrial transport, which is mediated by different mitochondrial membrane proteins (CPTs, CAT, and CACT) and by L-carnitine. Reduced levels of l-carnitine have been reported in NAFLD patients, which may contribute to reduced fatty acid oxidation and subsequent mitochondrial impairment and concomitant ROS production [54]. Supplementation with L-carnitine was shown to be associated with the improvement of liver inflammation and histological parameters in patients with NASH [55]. There is also evidence that L-carnitine supplementation ameliorates steatosis and improves mitochondrial function in the liver by increasing fatty acid oxidation in diabetic mice [56].
OxS can also lead to damage to macromolecules, resulting in the formation of toxic products. For example, lipid oxidation/peroxidation results in the formation of products such as malondialdehyde (MDA), lipid peroxides, 8-isoprostane, and 4-hydroxy-2-nonenal (4-HNE). These molecules are formed by hydroxyl radical attack to fatty acyl chains of phospholipids and triglycerides. Lipid peroxidation may lead to the downstream generation of reactive molecules (aldehydes) and/or the impairment of cellular structures and architecture, e.g., via structural changes in cellular membranes. In summary, OxS plays a pivotal role in NAFLD pathophysiology, which is linked to impaired lipid metabolism. Therefore, OxS and its downstream impairment of lipid metabolism are valid targets for NAFLD therapy.
3.2. Lipotoxicity in Non-Alcoholic Fatty Liver Disease
As already mentioned, OxS is linked to the pathogenesis of NAFLD, and this has been observed in experimental models of NAFLD/NASH [9] as well as in NAFLD patients who have an impaired redox balance, demonstrated by decreased levels of hepatic glutathione and anti-oxidant enzymes such as SOD and catalase [5][57]. The origin of the impaired redox status in NAFLD can be traced back to impaired lipid metabolism in the liver, resulting in an increased FFA pool in the hepatocytes [58]. This is due to an increased FFA uptake from dietary sources, increased de novo lipogenesis [59], and enhanced lipolysis in adipose tissue, which increases FFA delivery to the liver [60][61]. This excess FFA initially increases mitochondrial β-oxidation as an adaptive mechanism [53], which increases ROS production, but extramitochondrial oxidation is also enhanced [62], resulting in ROS overproduction, which then impairs the antioxidant balance and triggers lipotoxicity in the liver. Excess FFAs can also be incorporated as triglycerides in lipid droplets, causing steatosis [52]. Palmitate is the most abundant saturated long-chain fatty acid present in the diet, and its accumulation leads to increased levels of diacylglycerols (DAGs) and ceramides [63], both considered as lipotoxic lipid species, causing mitochondrial dysfunction and ER stress [48]. These toxic lipid species can induce lipotoxic effects via both direct and indirect mechanisms. For example, ceramides cause the impairment of mitochondrial function via inhibition of β-oxidation and increasing ROS production inside the mitochondria. In addition, DAG and ceramides activate several signalling pathways, including the proinflammatory NF-KB pathway and the NLRP3 inflammasome, as well as the JNK cell death signalling pathway [48]. These phenomena promote cellular dysfunction and further promote cell death and inflammation [64], in addition to impairing the antioxidant balance in hepatocytes. Altogether, mitochondria and ER contribute to the majority of cellular ROS production.
It has been demonstrated that FFA toxicity can be reduced by its incorporation into triglycerides and lipid droplets [65][66]. In fact, there is a growing body of evidence suggesting that hepatic TG accumulation is not itself harmful for hepatocytes, but rather a protective mechanism against lipotoxicity and consequently excessive ROS production [67][68]. Studies in experimental models of NAFLD also demonstrated that liver injury is caused by FFAs rather than TG accumulation. In fact, FFAs and their metabolites are known to induce hepatocyte injury by increasing OxS [69]. On the other hand, it has been demonstrated that not all species of FFAs are toxic to hepatocytes: MUFAs induce lipid droplet accumulation in vitro without affecting cell viability. In contrast, SFAs, such as palmitate, are toxic and induce only minimal changes in lipid droplet accumulation [70]. Therefore, saturated fatty acids are considered harmful to the cells compared to non-saturated fatty acids (e.g., MUFAs). In fact, the toxic effects of SFAs can be partially abolished by MUFAs, most likely through downregulation of proapoptotic pathways and favouring incorporation of SFAs into TG [64][70].
Parenchymal cells, e.g., hepatocytes, are the main cells affected by lipotoxicity-induced OxS in the liver. However, non-parenchymal cells (NPCs), including Kupffer cells, hepatic stellate cells (HSCs), and liver sinusoidal endothelial cells (LSECs), are also targets of OxS but may, compared with hepatocytes, respond in different ways [71]. Specifically, OxS promotes M1 polarization and activates inflammatory pathways in Kupffer cells, leading to an increased release of pro-inflammatory cytokines such as TNF-α. HSCs can be activated by lipid peroxidation and OxS, promoting their fibrogenic phenotype, resulting in increased synthesis of extracellular matrix components such as collagen [72]. Finally, OxS can damage LSECs. Thus, OxS induced by impaired lipid metabolism can directly kill cells (hepatocytes or LSECs), promote inflammation via activation of Kupffer cells, and promote fibrogenesis via activation of HSC. Extracellular vesicles and/or soluble factors from steatotic or injured hepatocytes/LSECs may aggravate the inflammatory response [73]. It is, therefore, important to also consider the role of NPCs in the pathogenesis of NAFLD and in the development of novel therapeutic targets.
It has been reported that the gut microbiota and the liver—gut axis play an important role in NAFLD [74]. This interaction is bi-directional: obesity and the increased dietary intake of (saturated) fats as well as the resulting changes in lipid metabolism cause profound changes in the composition of the gut microbiota [75]. On the other hand, changes in the microbiota can aggravate metabolic disturbances and NAFLD [76]. Dysbiosis is defined as changes in the microbiota that have detrimental effects. It has been demonstrated that the transfer of microbiota from obese to lean mice induces metabolic alterations in the recipient mice that are similar to those observed in the (obese) donor mice. It has also been demonstrated that mice with transferred gut microbiota from calorie-restricted mice show resistance to obesity and hepatic lipid accumulation when challenged with an HFD [77], which can be explained in part by the role of gut microbiota in the metabolism of PUFAs from dietary sources [78]. Dysbiosis induces changes in the synthesis of short chain fatty acids (scFFAs) by gut bacteria. These scFFAs act as ligands for G-protein coupled receptors (e.g., GPCR41 and GPCR43) that are involved in the pathogenesis of NAFLD. Dysbiosis also leads to decreased synthesis of Fasting-Induced Adipocyte Factor (FIAF). FIAF inhibits lipoprotein lipase, stimulating the release of free fatty acids [79]. The excess FFAs can subsequently disturb mitochondrial metabolism and lead to increased generation of ROS. Finally, dysbiosis increases intestinal permeability, leading to the translocation of bacteria to the liver, resulting in increased generation of ROS and exacerbating the inflammatory response [74]. Bacterial endotoxin or lipopolysaccharide (LPS) cause the activation of Kupffer cells through interaction with Toll-like receptors (e.g., TLR-4), which leads to the release of proinflammatory cytokines (e.g., TNFα) and free radicals [80]. Moreover, it has been demonstrated that palmitate induces inflammation and macrophage infiltration in the liver, and the subsequent palmitate-induced liver injury is exacerbated by a gut-derived endotoxin [81]. Since dysbiosis is strongly involved in the pathogenesis of NAFLD, the microbiota (microbiota transfer) may also be considered as a target in the therapy of NAFLD.
References
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124.
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med. Cell Longev. 2018, 2018, 9547613.
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801.
- Madan, K.; Bhardwaj, P.; Thareja, S.; Gupta, S.D.; Saraya, A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 2006, 40, 930–935.
- Palmieri, V.O.; Grattagliano, I.; Portincasa, P.; Palasciano, G. Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J. Nutr. 2006, 136, 3022–3026.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Medina, J.; Moreno-Otero, R. Pathophysiological basis for antioxidant therapy in chronic liver disease. Drugs 2005, 65, 2445–2461.
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091.
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26.
- Abe, Y.; Hines, I.N.; Zibari, G.; Pavlick, K.; Gray, L.; Kitagawa, Y.; Grisham, M.B. Mouse model of liver ischemia and reperfusion injury: Method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic. Biol. Med. 2009, 46, 1–7.
- Al-Asmari, A.K.; Khan, A.Q.; Al-Masri, N. Mitigation of 5-fluorouracil-induced liver damage in rats by Vitamin C via targeting redox-sensitive transcription factors. Hum. Exp. Toxicol. 2016, 35, 1203–1213.
- Smolková, K.; Mikó, E.; Kovács, T.; Leguina-Ruzzi, A.; Sipos, A.; Bai, P. Nuclear factor erythroid 2-related factor 2 in regulating cancer metabolism. Antioxid. Redox Signal. 2020, 33, 966–997.
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218.
- Kwak, T.W.K.Mi.; Itoh, K.; Yamamoto, M. Enhanced Expression of the Transcription Factor Nrf2 by Cancer Chemopreventive Agents: Role of Antioxidant Response Element-Like Sequences in the nrf2 Promoter. Mol. Cell. Biol. 2002, 22, 2883–2892.
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198.
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of NRF2 in liver diseases: Molecular, pharmacological, and epigenetic aspects. Antioxidants 2020, 9, 1–23.
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bionergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–70.
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424.
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031.
- Tanaka, Y.; Aleksunes, L.M.; Yeager, R.L.; Gyamfi, M.A.; Esterly, N.; Guo, G.L.; Klaassen, C.D. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharm. Exp. Ther. 2008, 325, 655–664.
- Zhang, Y.-K.J.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharm. 2012, 264, 305–314.
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1211–G1221.
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. Biomed Res. Int. 2015, 2015.
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med.2010, 48, 357–371.
- Zhang, Y.K.J.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharm. 2010, 245, 326–334.
- Sun, B.; Karin, M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016.
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115.
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86.
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-κB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243.
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB:p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta BBA 2008, 1783, 713–727.
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The Absence of Nrf2 Enhances NF-kB-Dependent Inflammation following Scratch Injury in Mouse Primary Cultured Astrocytes. Mediat. Inflamm. 2012, 2021, 1–9.
- Song, J.; Zhang, W.; Wang, J.; Yang, H.; Zhao, X.; Zhou, Q.; Wang, H.; Li, L.; Du, G. Activation of Nrf2 signaling by salvianolic acid C attenuates NF‑κB mediated inflammatory response both in vivo and in vitro. Int. Immunopharmacol. 2018, 63, 299–310.
- Zhang, D.M.; Guo, Z.X.; Zhao, Y.L.; Wang, Q.J.; Gao, Y.S.; Yu, T.; Chen, Y.K.; Chen, X.M.; Wang, G.Q. L-carnitine regulated Nrf2/Keap1 activation in vitro and in vivo and protected oxidized fish oil-induced inflammation response by inhibiting the NF-κB signaling pathway in Rhynchocypris lagowski Dybowski. Fish Shellfish Immunol. 2019, 93, 1100–1110.
- Liu, Z.; Dou, W.; Ni, Z.; Wen, Q.; Zhang, R.; Qin, M.; Wang, X.; Tang, H.; Cao, Y.; Wang, J.; et al. Deletion of Nrf2 leads to hepatic insulin resistance via the activation of NF-κB in mice fed a high-fat diet. Mol. Med. Rep. 2016, 14, 1323–1331.
- Yu, Q.; Xia, Z.; Liong, E.C.; Tipoe, G.L. Chronic aerobic exercise improves insulin sensitivity and modulates Nrf2 and NF‑κB/IκBα pathways in the skeletal muscle of rats fed with a high fat diet. Mol. Med. Rep.2019, 20, 4963–4972.
- Ou, Q.; Weng, Y.; Wang, S.; Zhao, Y.; Zhang, F.; Zhou, J.; Wu, X. Silybin Alleviates Hepatic Steatosis and Fibrosis in NASH Mice by Inhibiting Oxidative Stress and Involvement with the Nf-κB Pathway. Dig. Dis. Sci. 2018, 63, 3398–3408.
- Hodges, J.K.; Sasaki, G.Y.; Bruno, R.S. Anti-inflammatory activities of green tea catechins along the gut–liver axis in nonalcoholic fatty liver disease: Lessons learned from preclinical and human studies. J. Nutr. Biochem. 2020, 85, 108478.
- Damba, T.; Bourgonje, A.R.; Abdulle, A.E.; Pasch, A.; Sydor, S.; van den Berg, E.H.; Gansevoort, R.T.; Bakker, S.J.; Blokzijl, H.; Dullaart, R.P.; et al. Oxidative stress is associated with suspected non-alcoholic fatty liver disease and all-cause mortality in the general population. Liver Int. 2020, 40, 2148–2159.
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative Stress Enzymatic Antioxidant Status in Patients with Nonalcoholic Steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62.
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850.
- Coelho, J.M.; Cansanção, K.; de Mello Perez, R.; Leite, N.C.; Padilha, P.; Ramalho, A.; Peres, W. Association between serum and dietary antioxidant micronutrients and advanced liver fibrosis in non-alcoholic fatty liver disease: An observational study. PeerJ 2020, 8, 1–15.
- Hernandez, A.; Geng, Y.; Sepulveda, R.; Solis, N.; Torres, J.; Arab, J.P.; Barrera, F.; Cabrera, D.; Moshage, H.; Arrese, M. Chemical hypoxia induces pro-inflammatory signals in fat-laden hepatocytes and contributes to cellular crosstalk with Kupffer cells through extracellular vesicles. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165753.
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-κB signaling pathway. J. Gastroenterol. Hepatol. 2020, 35, 1998–2008.
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159.
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128.
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034.
- XPalomer; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab.2017, 29, 178–190.
- Fucho, R.; Casals, Ń.; Serra, D.; Herrero, L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J. 2017, 31, 1263–1272.
- Engin, A.B. What Is Lipotoxicity? In Obesity and Lipotoxicity, Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 197–220.
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932.
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Bloc’h, J.L.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283.
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Investig. 2015, 125, 4447–4462.
- Savic, D.; Hodson, L.; Neubauer, S.; Pavlides, M. The importance of the fatty acid transporter l-carnitine in non-alcoholic fatty liver disease (Nafld). Nutrients 2020, 12, 1–17.
- Malaguarnera, M.; Gargante, M.P.; Russo, C.; Antic, T.; Vacante, M.; Malaguarnera, M.; Avitabile, T.; Volti, G.L.; Galvano, F. L-carnitine supplementation to diet: A new tool in treatment of nonalcoholic steatohepatitisa randomized and controlled clinical trial. Am. J. Gastroenterol. 2010, 105, 1338–1345.
- Xia, Y.; Li, Q.; Zhong, W.; Dong, J.; Wang, Z.; Wang, C. L-carnitine ameliorated fatty liver in high-calorie diet/STZ-induced type 2 diabetic mice by improving mitochondrial function. Diabetol. Metab. Syndr.2010, 3, 1–10.
- VIDELA, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; Rojas, J. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268.
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 2009, 48, 1–26.
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735.
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351.
- Horst, K.W.T.; Serlie, M.J. Fructose Consumption, Lipogenesis,and Non-Alcoholic Fatty Liver Disease Enhanced Reader. Nutrients 2017, 9, 1–20.
- Cherkaoui-Malki, M.; Surapureddi, S.; el Hajj, H.I.; Vamecq, J.; Andreoletti, P. Hepatic Steatosis and Peroxisomal Fatty Acid Beta-oxidation. Curr. Drug Metab. 2012, 13, 1412–1421.
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902.
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451.
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082.
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1648–1655.
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting Triglyceride Synthesis Improves Hepatic Steatosis but Exacerbates Liver Damage and Fibrosis in Obese Mice with Nonalcoholic Steatohepatitis. Hepatology 2007, 45, 1366–1374.
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese Sr, R.V.; et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007, 6, 69–78.
- Li, S.; Hong, M.; Tan, H.Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxid. Med. Cell. Longev. 2016, 2016.
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-Coa desaturase. J. Biol. Chem. 2009, 284, 5637–5644.
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed Res. Int. 2016, 2016.
- Svegliati-Baroni, G.; Saccomanno, S.; van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver Int. 2001, 21, 1–12.
- Hernández, A.; Reyes, D.; Geng, Y.; Arab, J.P.; Cabrera, D.; Sepulveda, R.; Solis, N.; Buist-Homan, M.; Arrese, M.; Moshage, H. Extracellular vesicles derived from fat-laden hepatocytes undergoing chemical hypoxia promote a pro-fibrotic phenotype in hepatic stellate cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866,165857.
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, E1–E13.
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472.
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297.
- Wang, S.; Huang, M.; You, X.; Zhao, J.; Chen, L.; Wang, L.; Luo, Y.; Chen, Y. Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 2018, 8, 2–15.
- Miyamoto, J.; Igarashi, M.; Watanabe, K.; Karaki, S.I.; Mukouyama, H.; Kishino, S.; Li, X.; Ichimura, A.; Irie, J.; Sugimoto, Y.; et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 2019, 10, 1–5.
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339.
- Ji, Y.; Yin, Y.; Sun, L.; Zhang, W. The Molecular and Mechanistic Insights Based on Gut–Liver Axis: Nutritional Target for Non-Alcoholic Fatty Liver Disease (NAFLD) Improvement. Int. J. Mol. Sci. 2020, 21, 1–20.
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Fujita, K.; Yoneda, M.; Saito, S.; Saigusa, Y.; et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 1–14.


