 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Adriana Geisinger | + 3141 word(s) | 3141 | 2021-02-05 07:07:09 | | | |
| 2 | Dean Liu | -765 word(s) | 2376 | 2021-02-08 03:19:51 | | |
Video Upload Options
Flow cytometry has become an invaluable tool for the analysis of testicular heterogeneity, and for the purification of stage-specific spermatogenic cell populations, both for basic research and for clinical applications.
1. Biology of Spermatogenesis and Main Difficulties for Its Molecular Study
Mammalian spermatogenesis is the process of male gamete formation, a differentiation process that in normal conditions initiates at puberty and can last as long as the individual. It takes place in the testes, which bear at least seven somatic cell types, and at least 26 morphologically distinct germ cell stages[1].
Spermatogenesis consists of the successive occurrence of three main phases: (i) the proliferative phase of spermatogonia; (ii) the meiotic phase; and, (iii) the terminal differentiation phase or spermiogenesis (Figure 1). All three phases are essential for the reproductive health of the individuals, and consequently for the species survival.
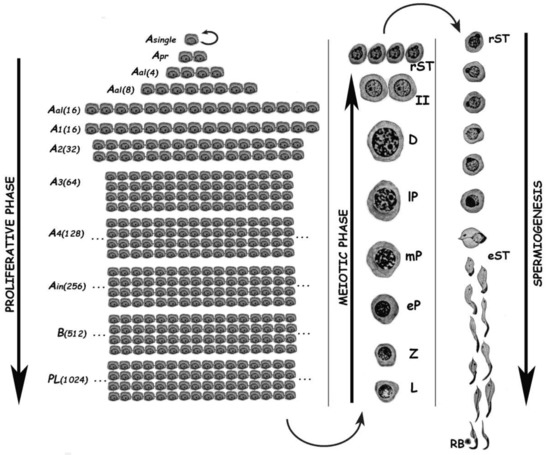
Figure 1. Schematic representation of the different germ cell stages along spermatogenesis of the rat. The three main phases of the spermatogenic process are indicated. Asingle, A single spermatogonium; Apr, A paired spermatogonia; Aal, A aligned spermatogonia; A1–A4, type A spermatogonia 1–4; Ain, intermediate spermatogonia; B, type B spermatogonia; PL, pre-leptotene spermatocytes; L, leptotene spermatocyte; Z, zygotene spermatocyte; eP, early pachytene spermatocyte; mP, medium pachytene spermatocyte; lP, late pachytene spermatocyte; D, diplotene spermatocyte; II, secondary spermatocyte; rST, round spermatids; eST, elongating spermatids; RB, residual body. In the proliferative phase, the numbers of spermatogonia derived from an A single are indicated in parenthesis.
The proliferative phase depends on the presence of spermatogonial stem cells (SSCs) that amplify themselves to maintain the SSC pool (self-renewal), but also give rise to progenitor spermatogonia (committed to differentiation). For instance, in the mouse SSCs are derived from gonocytes and reside in the population of Asingle (As) spermatogonia. The latter initiate mitotic proliferation either to produce new As spermatogonia by complete cytokinesis, or to give rise to chains of Apaired (Apr), and Aaligned (Aal) spermatogonia. These represent undifferentiated spermatogonia that are connected by intercellular bridges as a consequence of incomplete cytokinesis. Aal spermatogonia, as well as a few Apr spermatogonia, differentiate into A1 spermatogonia without division, and then proliferate mitotically five times to sequentially form A2, A3, A4, intermediate, and type B spermatogonia, collectively termed differentiated spermatogonia. Afterwards, type B spermatogonia divide into two primary diploid spermatocytes that enter meiosis[2].
In the meiotic phase, ploidy halving is accomplished through a single round of DNA replication followed by two cellular divisions. During the first meiotic division (meiosis I) homologous chromosomes segregate, and primary spermatocytes (4C, 2n) give rise to secondary ones with 2C DNA content but already haploid (1n). Secondary spermatocytes enter meiosis II, and separation of sister chromatids takes place, generating the round spermatids (1C, 1n), which initiate spermiogenesis. Notably, the reduction in ploidy is of fundamental importance for gametogenesis in all sexually reproducing organisms, as at the time of fertilization fusion of the male and female gametes leads to the restoration of the species chromosome number. In addition to its reductive nature, meiosis is also very peculiar regarding the exchanges of genetic material that take place between homologous chromosomes during prophase I. Homologous chromosomes (i.e., of maternal and paternal origin within each pair) align, and then synapse via a highly specialized proteinaceous structure—the synaptonemal complex (SC)—that assembles during prophase I, enabling the closeness required for homologous recombination[3][4]. Due to the importance of the unique events that take place during meiotic prophase I (formation of the SCs, alignment and pairing, recombination), this has been the most extensively studied meiotic stage. As it is a very long stage, it has been divided into different substages to facilitate its study: leptotene (L), zygotene (Z), pachytene (P), diplotene (D), and diakinesis (see Figure 1). The assembly of the SC starts in L, homologous pairing takes place in Z, and recombination (crossing-over) is the hallmark of P. During D, the SCs disassemble[5]. The eventual alteration of these events often leads to spermatogenic arrest and infertility[6][7][8][9].
Spermiogenesis is the third and final phase of spermatogenesis. Along this post-meiotic stage, round spermatids go through a series of profound morphological and functional changes, giving rise to mature spermatozoa (see Figure 1). In the mouse, spermatids can be morphologically classified as steps 1–8 round spermatids, and steps 9–16 elongating ones[10]. Particularly in the chromatin, the main change in spermatids is the replacement of most histones by transition proteins first, and then by protamines.
Cellular heterogeneity represents a major drawback for the identification of molecular factors, and the unveiling of molecular mechanisms underneath gamete formation. Studies seeking these goals usually require the isolation of cells from the developmental stage of interest. The lack of an effective in vitro culture system[11][12] has also hampered spermatogenic stage-specific molecular studies.
2. The Spermatogenic Process from a Flow Cytometric Perspective
As mentioned above, spermatogenesis comprises several stages from spermatogonia to elongated spermatids, which present a wide range of variation in their cellular size, shape, inner complexity, chromatin structure, and DNA content. Most these parameters can be detected and measure-assigned by FCM, thus explaining why this powerful technology came to stay in this research area[13]. Table 1 summarizes some important features of the most relevant testicular cell types that are useful in FCM.
Table 1. Principal characteristics of testicular cells from adult rodents.
| Approximate Cellular Size (µm) | Cellular/Nuclear Shape | Inner Complexity | Chromatine Structure | DNA Content | |
|---|---|---|---|---|---|
| Type A spermatogonia | 12–14 | Ovoid nuclei | Very low | Homogenous euchromatin | 2C-4C * |
| Type B spermatogonia | 8–10 | Round nuclei | Low | Heterochromatin along nuclear periphery | 2C-4C * |
| PreLeptotene spermatocytes | 7.6–8.2 | Round nuclei | Low | Heterochromatin along nuclear periphery | 2C-4C * |
| Leptotene spermatocytes | 8–10 | Round nuclei | Low | Condensed chromosomes forming thin filaments | 4C |
| Zygotene spermatocytes | 10–12 | Round nuclei | Medium | Clumps of dense chromatin, mainly at the nuclear periphery | 4C |
| Pachytene spermatocytes | 12–18 | Large round nuclei; thin rim of cytoplasm; cell volume increases along the stage | High | Abundant clumps of dense chromatin; shorter and thicker chromosomes | 4C |
| Round spermatids | 10 | Round cells with round nuclei | Very low | Homogenous euchromatin; densely stained chromocenter in the middle | 1C |
| Elongating/elongated spermatids | 4–8 | Sickle-shaped nuclei | Low-Medium | Increasingly compacted chromatin | 1C |
| Leydig cells | 10–12 | Polyhedral cells with eccentrically located ovoid nuclei; abundant citoplasmic lipid droplets. | Very high | Prominent nucleoli; abundant peripheral heterochromatin | 2C |
| Sertoli cells | Height (from basal to apical surface): 90; Base: 30 | Columnar, Irregular, with apical and lateral invaginations; oval-shaped nuclei | High | Dark nucleolus; central condensation area | 2C |
Information was extracted from references[14][15], and our own experience. * 4C DNA content in these cells corresponds to the G2 cell cycle phase. Inner complexity level assignment was performed based mainly on observation of flow cytometry (FCM) side scatter parameter.
Regarding DNA content, testicular cell suspensions can be stained with a fluorescent dye and analyzed by FCM. The choice of dyes that stoichiometrically bind DNA allows the discrimination of cell populations with different ploidy based on their fluorescence intensity. In the spermatogenic context, with many cell types differing in their DNA content, this sole parameter enables a gross first simplification concerning testis heterogeneity. Three main groups of events can be distinguished: C (round spermatids, elongating spermatids), 2C (several types of G1 spermatogonia, secondary spermatocytes, testicular somatic cells), and 4C (different stages of primary spermatocytes, G2 spermatogonia) (see the histogram in Figure 2B). In adult specimens, a fourth population (elongated spermatids, sperm) can be observed, bearing an apparently sub-haploid DNA content[16]. Different fluorochromes with distinct action mechanisms have been employed for the DNA content-based discrimination of testicular cell populations (for a revision, see[13]). When the aim is to classify cells for downstream molecular applications, then the vital dye Hoechst 33342 (Ho342) has been the most commonly used dye[17][18][19][20][21][22][23][24].Ho342 is excited in the UV range and emits in blue and red; a combination of both fluorescences enables the resolution of various testicular cell populations (Figure 2A).On the other hand, we have developed an alternative sorting protocol for testicular cell populations based on the vital dye Vybrant DyeCycle Green (VDG) (Figure 2B). As VDG is excited with a blue laser (thus avoiding the potential damage to nucleic acids caused by UV light), it represents an advantage for downstream applications where nucleic acid integrity is an important issue[25][26].
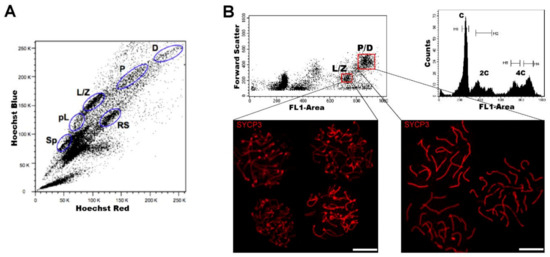
Figure 2. FCM analysis of mouse testicular cell suspensions employing two different DNA dyes. (A) Representative Hoechst 33342 (Ho342) FCM profile obtained from the analysis of adult mouse testis, based on emitted blue and red fluorescence. The various spermatogenic cell populations that can be distinguished, and are indicated, are: spermatogonia (Sp), pre-leptotene (pL), leptotene–zygotene (L/Z), pachytene (P), diplotene (D), and round spermatids (RS). This image is reproduced from reference[27] with permission of J. Vis. Exp. (B) Representative FCM profiles from the analysis of a 22-day-old mouse testicular cell suspension stained with Vybrant DyeCycle Green (VDG). A Forward Scatter vs. FL1-Area (VDG fluorescence intensity) dot plot and its corresponding histogram are shown. Histogram peaks corresponding to C, 2C and 4C cell populations, and sorting gates within the 4C population (delimited in red) in the dot plot, are indicated. Note the absence of the apparently sub-haploid peak in the histogram, as the profile is from a juvenile animal. Examples of immunolocalizations of SYCP3 (synaptonemal complex (SC) protein 3, a lateral-element–SC component) on cellular spreads from sorted cells, in order to confirm the purity of the sorted fractions, are shown below. As can be seen, cells coming from the selected gates correspond to different stages of the first meiotic prophase: L/Z, in which simple axes (L) and short stretches of SCs (Z) are present; or P/D with completely assembled (P), or disassembling (D) SCs. Bars correspond to 10 µm.
The combination of these three parameters gives rise to multi-parametric analyses with higher information input than when considering just one, thus enabling an advance of one more step towards the distinction of different cell types and stages[13].
Where certain cell types cannot be solely identified or purified based on ploidy, different strategies have been employed such as the introduction of fluorescent labels [e.g. 97,99]. Specific antibody-dependent identification and sorting of cell types has been also a very common strategy for the identification and isolation of cell types that are hardly distinguishable or not distinguishable at all in the cytometric profiles, such as specific populations of spermatogonia, or certain somatic cell types [e.g. 100].
3. FCM as an Analytical Tool of Spermatogenesis
A well-recognized advantage of FCM is its high quantitative analytical power and statistical weight. Depending on the equipment, hundreds or even thousands of events per second can be analyzed. Reproductive biologists have harnessed the analytical power of FCM for multi-parametric analyses of testicular cell suspensions. They have taken advantage of the various differences between cell types and stages, which translated to FCM profiles, allow the assessment of spermatogenic advance. Multi-parametric FCM analyses have been historically employed to study postnatal development of the male gonad in different species [28][29][30][31][32][33][34][35][36][37][38][39][40].
FCM profiling of testicular cell suspensions has also proved useful in the diagnosis of human male infertility[41], in assessing sperm quality[42], in the study of testicular cancer[43], and in the characterization of reproductive disease mouse models [44], among other analytical applications.
4. FCM as a Preparative Tool in Spermatogenic Studies
Whenever the available FCM equipment is also a sorter, any well-defined population in the dot plots can be chosen and classified by fluorescence activated cell sorting (FACS) for downstream studies. The strategies for the distinction and subsequent sorting of spermatogenic cell populations have been varied, ranging from multiparametric analyses that rely on differences in DNA content and light scattering, to the employment of specific antibodies against the stages of interest.
FCM allows the discrimination of a higher number of spermatogenic cell types and enables the obtainment of highly pure cell populations via FACS[28][45]; therefore, it presents important advantages for transcriptomic studies. Therefore, flow-sorted spermatogenic cell populations have been employed for the obtainment of highly reliable transcriptomic profiles, including scarce cell populations that are hard to obtain with other methods, such as early meiotic prophase stages[17][46][47]. FACS has also complemented single-cell approaches in various studies, either for the enrichment in certain cell types[23][48][49][50][51], for the study of different types of spermatogonia[48][52][53][54][55], or as a way to complement random cell picking (unsorted) for more accurate cell-type assignment [23][56]. FACS has been also combined with the synchronization of spermatogenesis[20][49].
FACS-sorted spermatogenic cell populations have been also employed in studies addressing the understanding of chromatin accessibility, histone modifications, DNA methylomes, and three-dimensional (3D) chromatin structure, during mouse and/or
human spermatogenesis[18][21][22][24][57][58] .
In addition, in vitro recapitulation of the spermatogenic process has been a precious goal among reproductive biologists for at least six decades, as it would facilitate, among others, studies on the requirements of the process in a controlled in vitro environment; research that is difficult and/or unethical to perform directly in vivo; studies on the molecular mechanisms of
pathologies such as testicular cancer or male infertility; fertility restoration or preservation, by the production of haploid male germ cells from undifferentiated germ cells isolated from infertile adult patients, or from pre-pubertal cancer patients before the application of gonadotoxic treatments, respectively[11][59][60][61]. As SSCs are responsible for the continuous production of spermatogonia that sustain spermatogenesis [62], the generation of in vitro culture systems to efficiently maintain and expand SSCs is fundamental for progress towards the above-mentioned goals. FACS, based on specific markers, presents important advantages for the obtainment of high purity SSCs, as it provides morphological data of cells, allows the simultaneous detection of multiple surface markers, and is informative on eventual quantitative differences of biochemical markers between cellular subpopulations [e.g. [51][63][64][65][66][67][68].
5. Conclusions and Perspectives
Being such a heterogeneous tissue, mammalian testis has represented a challenge in the attempt to unravel the molecular bases of spermatogenesis. FCM has significantly contributed to the field in allowing to identify and eventually purify a high number of specific cell types. As more advances are developed, we foresee that the FCM has a lot more to contribute in this area of knowledge, as there is still a lot of ground for potential complementation between FCM and complementary approaches.
References
- Hess, R.A.; de Franca, L.R. Spermatogenesis and cycle of the seminiferous epithelium. Adv. Exp. Med. Biol. 2008, 636, 1–15.
- De Rooij, D.G.; Griswold, D.K.M. Questions about spermatogonia posed and answered since 2000. J. Androl. 2012, 33, 1085–1995.
- Fraune, J.; Schramm, S.; Alsheimer, M.; Benavente, R. The mammalian synaptonemal complex: Protein components, assembly and role in meiotic recombination. Exp. Cell Res. 2012, 318, 1340–1346.
- Bolcun-Filas, E.; Ann Handel, M. Meiosis: The chromosomal foundation of reproduction. Biol. Reprod. 2018, 99, 112–126.
- Cobb, J.; Handel, M.A. Dynamics of meiotic prophase I during spermatogenesis: From pairing to division. Semin. Cell Dev. Biol. 1998, 9, 445–450.
- Handel, M.A.; Schimenti, J.C. Genetics of mammalian meiosis: Regulation, dynamics and impact on fertility. Nat. Rev. Genet. 2010, 11, 124–136.
- Hann, M.C.; Lau, P.E.; Tempest, H.G. Meiotic recombination and male infertility: From basic science to clinical reality? Asian J. Androl. 2011, 13, 212–218.
- Geisinger, A.; Benavente, R. Mutations in genes coding for synaptonemal complex proteins and their impact on human fertility. Cytogenet. Genome Res. 2016, 150, 77–85.
- Gheldof, A.; Mackay, D.J.G.; Cheong, Y.; Verpoest, W. Genetic diagnosis of subfertility: The impact of meiosis and maternal effects. J. Med. Genet. 2019, 56, 271–282.
- Clermont, Y. Kinetics of spermatogenesis in mammals: Seminiferous epithelium cycle and spermatogonial renewal. Physiol. Rev. 1972, 52, 198–236.
- Handel, M.A.; Eppig, J.J.; Schimenti, J.C. Applying “Gold Standards” to in vitro-derived germ cells. Cell 2014, 157, 1257–1261.
- Komeya, M.; Sato, T.; Ogawa, T. In vitro spermatogenesis: A century-long research journey, still half way around. Reprod. Med. Biol. 2018, 17, 407–420.
- Geisinger, A.; Rodríguez-Casuriaga, R. Flow cytometry for gene expression studies in mammalian spermatogenesis. Cytogenet. Genome Res. 2010, 128, 46–56.
- Bellvé, A.R. Purification, culture, and fractionation of spermatogenic cells. Methods Enzymol. 1993, 225, 84–113.
- Wong, V.; Russell, L.D. Three-dimensional reconstruction of a rat stage V Sertoli cell: I. Methods, basic configuration, and dimensions. Am. J. Anat. 1983, 167, 143–161.
- Spano, M.; Evenson, D.P. Flow cytometric analysis for reproductive biology. Biol. Cell 1993, 78, 53–62.
- Fallahi, M.; Getun, I.V.; Wu, Z.K.; Bois, P.R.J. A global expression switch marks pachytene initiation during mouse male meiosis. Genes 2010, 1, 469–483.
- Getun, I.V.; Wu, Z.K.; Khalil, A.M.; Bois, P.R.J. Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep. 2010, 11, 555–560.
- Zhu, Z.; Li, C.; Yang, S.; Tian, R.; Wang, J.; Yuan, Q.; Dong, H.; He, Z.; Wang, S.; Li, Z. Dynamics of the transcriptome during human spermatogenesis: Predicting the potential key genes regulating male gametes generation. Sci. Rep. 2016, 6, 19069.
- Chen, Y.; Zheng, Y.; Gao, Y.; Lin, Z.; Yang, S.; Wang, T.; Wang, Q.; Xie, N.; Hua, R.; Liu, M.; et al. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018, 28, 879–896.
- Gaysinskaya, V.; Miller, B.F.; De Luca, C.; van der Heijden, G.W.; Hansen, K.D.; Bortvin, A. Transient reduction of DNA methylation at the onset of meiosis in male mice. Epigenetics Chromatin 2018, 11, 15.
- Ernst, C.; Eling, N.; Martinez-Jimenez, C.P.; Marioni, J.C.; Odom, D.T. Staged developmental mapping and X chromosome transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019, 10, 1251.
- Jung, M.; Wells, D.; Rusch, J.; Ahmed, S.; Marchini, J.; Myers, S.; Conrad, D.F. Unified single-cell analysis of testis gene regulation and pathology in 5 mouse strains. eLife 2019, 8, e43966.
- Vara, C.; Paytuvi-Gallart, A.; Cuartero, Y.; Le Dily, F.; Garcia, F.; Salva-Castro, J.; Gómez, H.L.; Julià, E.; Moutinho, C.; Aiese Cigliano, R.; et al. Three-dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 2019, 28, 352–367.
- Rodríguez-Casuriaga, R.; Santiñaque, F.F.; Folle, G.A.; Souza, E.; López-Carro, B.; Geisinger, A. Rapid preparation of rodent testicular cell suspensions and spermatogenic stages purification by flow cytometry using a novel blue laser-excitable vital dye. Methods X 2014, 1, 239–243.
- Geisinger, A.; Rodríguez-Casuriaga, R. Flow cytometry for the isolation and characterization of rodent meiocytes. Methods Mol. Biol. 2017, 1471, 217–230.
- Getun, I.V.; Torres, B.; Bois, P.R. Flow cytometry purification of mouse meiotic cells. J. Vis. Exp. 2011, 50, 2602.
- Malkov, M.; Fisher, Y.; Don, J. Developmental schedule of the postnatal rat testis determined by flow cytometry. Biol. Reprod. 1998, 59, 84–92.
- Rodríguez-Casuriaga, R.; Geisinger, A.; Santiñaque, F.F.; López-Carro, B.; Folle, G.A. High-purity flow sorting of early meiocytes based on DNA analysis of guinea pig spermatogenic cells. Cytometry A 2011, 79, 625–634.
- Janca, F.C.; Jost, L.K.; Evenson, D.P. Mouse testicular and sperm cell development characterized from birth to adulthood by dual parameter flow cytometry. Biol. Reprod. 1986, 34, 613–623.
- Petit, J.M.; Ratinaud, M.H.; Cordelli, E.; Spano, M.; Julien, R. Mouse testis cell sorting according to DNA and mitochondrial changes during spermatogenesis. Cytometry 1995, 19, 304–312.
- Suter, L.; Koch, E.; Bechter, R.; Bobadilla, M. Three-parameter flow cytometric analysis of rat spermatogenesis. Cytometry 1997, 27, 161–168.
- Vigodner, M.; Lewin, L.M.; Shochat, L.; Mittelman, L.; Golan, R. Meiosis in the golden hamster: A confocal microscopy and flow cytometric analysis. Molec. Reprod. Dev. 2003, 64, 86–95.
- Vigodner, M.; Lewy, H.; Lewin, L.M.; Shochat, L.; Golan, R. Evidence for biological rhythm in spermatogenesis in the pubertal hamster (Mesocricetus auratus): A flow cytometric study. Life Sci. 2004, 74, 1119–1126.
- Neubauer, K.; Jewgenow, K.; Blottner, S.; Wildt, D.E.; Pukazhenthi, B.S. Quantity rather than quality in teratospermic males: A histomorphometric and flow cytometric evaluation of spermatogenesis in the domestic cat (Felis catus). Biol. Reprod. 2004, 71, 1517–1524.
- Oskam, I.C.; Ropstad, E.; Andersen Berg, K.; Fredriksen, B.; Larsen, S.; Dahl, E.; Andresen, O. Testicular germ cell development in relation to 5alpha-androstenone levels in pubertal entire male pigs. Theriogenology 2008, 69, 967–976.
- Aslam, H.; Schneiders, A.; Perret, M.; Weinbauer, G.F.; Hodges, J.K. Quantitative assessment of testicular germ cell production and kinematic and morphometric parameters of ejaculated spermatozoa in the grey lemur, Microebus murinus. Reproduction 2002, 123, 323–332.
- Wistuba, J.; Schrod, A.; Greve, B.; Hodges, J.K.; Aslam, H.; Weinbauer, G.F.; Luetjens, M. Organization of seminiferous epithelium in primates: Relationship to spermatogenic efficiency, phylogeny, and mating system. Biol. Reprod. 2003, 69, 582–591.
- Blanchard, Y.; Lavault, M.T.; Quernee, D.; Le Lannou, D.; Lobel, B.; Lescoat, D. Preparation of spermatogenic cell populations at specific stages of differentiation in the human. Mol. Reprod. Dev. 1991, 30, 275–282.
- Hittmair, A.; Rogatsch, H.; Offner, F.; Feichtinger, H.; Ofner, D.; Mikuz, G. Deoxyribonucleic acid flow cytometry and semiquantitative histology of spermatogenesis: A comparative study. Fertil. Steril. 1992, 58, 1040–1045.
- Baksi, A.; Vasan, S.S.; Dighe, R.R. DNA Flow cytometric analysis of the human testicular tissues to investigate the status of spermatogenesis in azoospermic patients. Sci. Rep. 2018, 8, 11117.
- Farah, O.I.; Cuiling, L.; Jiaojiao, W.; Huiping, Z. Use of fluorescent dyes for readily recognizing sperm damage. J. Reprod. Infertil. 2013, 14, 120–125.
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Stanton, M.; Slack, J.; Andrews, P.; Pagliaro, L.; Bryce, A.H. Clonal analyses of refractory testicular germ cell tumors. PLoS ONE 2019, 14, e0213815.
- Hernández-López, D.; Geisinger, A.; Trovero, M.F.; Santiñaque, F.F.; Brauer, M.; Folle, G.A.; Benavente, R.; Rodríguez-Casuriaga, R. Familial primary ovarian failure associated with a SYCE1 point mutation: Defective meiosis elucidated in humanized mice. Molec. Hum. Reprod. 2020, 26, 485–497.
- Bastos, H.; Lassalle, B.; Chicheportiche, A.; Riou, L.; Testart, J.; Allemand, I.; Fouchet, P. Flow cytometric characterization of viable meiotic and postmeiotic cells by Hoechst 33342 in mouse spermatogenesis. Cytometry A 2005, 65A, 40–49.
- Da Cruz, I.; Rodríguez-Casuriaga, R.; Santiñaque, F.F.; Farías, J.; Curti, G.; Capoano, C.A.; Folle, G.A.; Benavente, R.; Sotelo-Silveira, J.R.; Geisinger, A. Transcriptome analysis of highly purified mouse spermatogenic cell populations: Gene expression signatures switch from meiotic-to-postmeiotic-related processes at pachytene stage. BMC Genom. 2016, 17, 294.
- Trovero, M.F.; Rodríguez-Casuriaga, R.; Romeo, C.; Santiñaque, F.F.; François, M.; Folle, G.A.; Benavente, R.; Sotelo-Silveira, J.R.; Geisinger, A. Revealing stage-specific expression patterns of long noncoding RNAs along mouse spermatogenesis. RNA Biol. 2020, 17, 350–365.
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernströer, B.; et al. The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep. 2018, 25, 1650–1667.
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; et al. A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-Seq. Dev. Cell 2018, 46, 651–667.
- Grive, K.J.; Hu, Y.; Shu, E.; Grimson, A.; Elemento, O.; Grenier, J.K.; Cohen, P.E. Dynamic transcriptome profiles within spermatogonial and spermatocyte populations during postnatal testis maturation revealed by single-cell sequencing. PLoS Genet. 2019, 15, e1007810.
- La, H.M.; Mäkelä, J.A.; Chan, A.L.; Rossello, F.J.; Nefzger, C.M.; Legrand, J.M.D.; De Seram, M.; Polo, J.M.; Hobbs, R.M. Identification of dynamic undifferentiated cell states within the male germline. Nat. Commun. 2018, 9, 2819.
- Song, H.W.; Bettegowda, A.; Lake, B.B.; Zhao, A.H.; Skarbrevik, D.; Babajanian, E.; Sukhwani, M.; Shum, E.Y.; Phan, M.H.; Plank, T.M.; et al. The homeobox transcription factor RHOX10 drives mouse spermatogonial stem cell establishment. Cell Rep. 2016, 17, 149–164.
- Liao, J.; Ng, S.H.; Luk, A.C.; Suen, H.C.; Qian, Y.; Lee, A.W.T.; Tu, J.; Fung, J.C.L.; Tang, N.L.S.; Feng, B.; et al. Revealing cellular and molecular transitions in neonatal germ cell differentiation using single cell RNA sequencing. Development 2019, 146, dev174953.
- Makino, Y.; Jensen, N.H.; Yokota, N.; Rossner, M.J.; Akiyama, H.; Shirahige, K.; Okada, Y. Single cell RNA-sequencing identified Dec2 as a suppressive factor for spermatogonial differentiation by inhibiting Sohlh1 expression. Sci. Rep. 2019, 9, 6063.
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-cell RNA-seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell 2017, 20, 858–873.
- Wang, M.; Liu, X.; Chang, G.; Chen, Y.; An, G.; Yan, L.; Gao, S.; Xu, Y.; Cui, Y.; Dong, J.; et al. Single-Cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis. Cell Stem Cell 2018, 23, 599–614.
- Patel, L.; Kang, R.; Rosenberg, S.C.; Qiu, Y.; Raviram, R.; Chee, S.; Hu, R.; Ren, B.; Cole, F.; Corbett, K.D. Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat. Struct. Mol. Biol. 2019, 26, 164–174.
- Lesch, B.J.; Dokshin, G.A.; Young, R.A.; McCarrey, J.R.; Page, D.C. A set of genes critical to development is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. Proc. Natl. Acad. Sci. USA. 2013, 110, 16061–16066.
- Hayashi, K. In vitro reconstitution of germ cell development. Biol. Reprod. 2019, 101, 567–578.
- Oliver, E.; Stukenborg, J.B. Rebuilding the human testis in vitro. Andrology 2020, 8, 825–834.
- Pelzman, D.L.; Orwig, K.E.; Hwang, K. Progress in translational reproductive science: Testicular tissue transplantation and in vitro spermatogenesis. Fertil. Steril. 2020, 113, 500–509.
- Phillips, B.T.; Gassei, K.; Orwig, K.E. Spermatogonial stem cell regulation and spermatogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1663–1678.
- Valli, H.; Sukhwani, M.; Dovey, S.L.; Peters, K.A.; Donohue, J.; Castro, C.A.; Chu, T.; Marshall, G.R.; Orwig, K.E. Fluorescence and magnetic-activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertil. Steril. 2014, 102, 566–580.
- Tan, K.; Wilkinson, M.F. A single-cell view of spermatogonial stem cells. Curr. Opin. Cell Biol. 2020, 67, 71–78.
- Law, N.C.; Oatley, M.J.; Oatley, J.M. Developmental kinetics and transcriptome dynamics of stem cell specification in the spermatogenic lineage. Nat. Commun. 2019, 10, 2787.
- Nakagawa, T.; Sharma, M.; Nabeshima, Y.; Braun, R.E.; Yoshida, S. Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment. Science 2010, 328, 62–67.
- Velte, E.K.; Niedenberger, B.A.; Serra, N.D.; Singh, A.; Roa-DeLaCruz, L.; Hermann, B.P.; Geyer, C.B. Differential RA responsiveness directs formation of functionally distinct spermatogonial populations at the initiation of spermatogenesis in the mouse. Development 2019, 146, dev173088.
- Sohni, A.; Tan, K.; Song, H.W.; Burow, D.; de Rooij, D.G.; Laurent, L.; Hsieh, T.C.; Rabah, R.; Hammoud, S.S.; Vicini, E.; et al. The neonatal and adult human testis defined at the single-cell level. Cell Rep. 2019, 26, 1501–1517.


