 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luca Mologni | + 6224 word(s) | 6224 | 2021-02-01 06:08:33 | | | |
| 2 | Peter Tang | Meta information modification | 6224 | 2021-02-21 09:33:49 | | |
Video Upload Options
Pharmacological cancer therapy is often based on the concurrent inhibition of different survival pathways to improve treatment outcomes and to reduce the risk of relapses. While this strategy is traditionally pursued only through the co-administration of several drugs, the recent development of multi-targeting drugs (i.e., compounds intrinsically able to simultaneously target several macromolecules involved in cancer onset) has had a dramatic impact on cancer treatment.
1. Introduction
One of the most spectacular breakthroughs in cancer therapy in the past 20 years is undoubtedly represented by the introduction of imatinib mesylate for the treatment of Philadelphia-positive (Ph+) leukemia [1][2]. The success of imatinib instilled the idea that most tumors could be treated by a single, targeted drug that inhibits the main driver of transformation. However, the initial enthusiasm subsided when it became clear that, on the contrary, most tumors harbor at least two or more driver lesions, all of which need to be targeted to achieve disease control [3]. Even when one major driver can be identified, cells can often switch to alternative signaling to resist therapy [4] (Figure 1). Therefore, dual targeting is being intensively investigated to improve treatment outcomes [5][6][7]. The rationale behind the better anticancer activity of combined inhibition is based on (i) complete and simultaneous suppression of cooperating oncogenic signals and (ii) concurrent block of bypass signaling tracks that may mediate drug resistance. Obviously, inhibiting only one of two cooperating oncogenic signals will produce a partial effect. Moreover, a cancer cell is less likely to develop resistance to two drugs than to one (probability of dual resistance is the product of the probabilities of single resistance). In this scenario, dually targeted molecules represent a further step: They achieve combined targeting without the unwanted combination of side effects that can be observed in traditional dual targeting by two different molecules. From a medicinal chemistry point of view, “dual” may have different meanings: researchers can develop a dual inhibitor by intentionally aiming at two specific targets or simply by exploiting off-target activity of a compound primarily targeted to another kinase. As drugs are most often not mono-specific, off-targets can be detrimental, beneficial, or irrelevant to the disease under study. Thus, in general, “dual” indicates a drug that targets two relevant kinases at comparable potency, showing some selectivity over other unwanted targets.
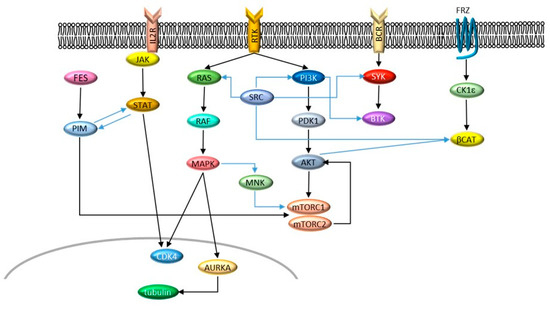
Figure 1. Signaling networks described in this entry. Cross-talks are indicated by blue arrows.
2. Dual Kinase Targeting in AML
Acute myeloid leukemia (AML) is a cancer defined by the infiltration of bone marrow, blood, and other tissues by proliferative, clonal, abnormally differentiated and poorly differentiated cells of the hematopoietic system [8]. AML is characterized by cytogenetic and molecular heterogeneity; genetic abnormalities include amplifications, deletions, rearrangements, and point mutations [8][9]. The last WHO classification has added RUNX1 mutations and BCR-ABL1 rearrangements to the previous main molecular markers (NPM1, CEBPA, and FLT3) [10]. Most AML patients respond well to chemotherapy, but subsequent relapse is common, and the prognosis is still poor, with approximately 30% of adult patients under 60 years of age achieving 5-year survival and a median survival of 5 to 10 months for older patients [11][12][13].
New, more targeted therapeutic approaches have been attempted to meet this clinical need, such as the use of fms-like tyrosine kinase 3 (FLT3) inhibitor midostaurin [14][15], isocitrate dehydrogenase-2 (IDH2) inhibitor enasidenib [16], and calicheamicin conjugated anti-CD33-antibody gemtuzumab [17]. As commonly observed, the major limitation of targeting these individual oncogenes is the development of resistance; hence, long-term remissions in AML still require the combined use of cytotoxic chemotherapy and/or targeted agents along with stem cell transplant, with ensuing systemic toxicity [11]. The dual inhibition of protein kinases in AML has been extensively explored to achieve response to relapsing and resistant tumors avoiding toxic side effects, mainly involving co-inhibition of the phosphatidylinositol 3-kinase(PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) (PI3K/AKT/mTOR) pathway and FLT3 kinase.
In hematopoietic cells, the PI3K/AKT/mTOR pathway is an important regulator of cell growth, proliferation, and survival; the amplification and deregulation of this pathway confer resistance to apoptosis, growth inhibition, and differentiation on leukemia cells [18]. Constitutive activation of the PI3K/AKT/mTOR signal is common in AML patients and linked to reduced survival, making the targeting of its components a prime strategy for the treatment of AML [11][19]. The PI3K family of lipid kinases comprises several regulatory and catalytic subunits encoded by different genes [20]. PI3K is the first enzyme along the PI3K/AKT/mTOR pathway to be activated by receptor tyrosine kinases (RTKs) or through an adaptor molecule; the main group of PI3Ks involved in neoplastic diseases is class I PI3K [18]. The mTOR protein is involved in two major complexes: the mTOR complex 1 (mTORC1) and the mTOR complex 2 (mTORC2). The mTORC1 controls major regulators of cellular growth and proliferation like the cyclin-dependent kinase inhibitor p27kip1, the retinoblastoma protein (Rb), cyclin D1, c-myc, and STAT3, while the mTORC2 complex has a role in regulating AKT and in resistance to apoptosis [21]. The mTOR pathway helps regulate several key cellular processes, including cell growth, proliferation and cycling, and protein synthesis. In AML, dysregulation of these processes aids leukemia cells in enhancing growth and proliferation and resisting apoptosis; increased mTOR activity thus plays a role in AML relapse and initiation [21][22]. The first mTOR inhibitor discovered was rapamycin; rapamycin and its analogs (rapalogs) bind specifically to the FKBP12-rapamycin-binding (FRB) domain of mTOR, preventing the formation of the mTORC1 complex and are thus mTORC1- specific [23]. While preclinical studies on mTORC1 inhibitors have shown that rapamycin and its analogs are able to slow the growth and proliferation of AML cell lines and leukemic blasts [24], in numerous clinical studies on AML patients, they have shown a very limited efficacy when used as monotherapies [11], and even their combination with conventional chemotherapies did not yield a clear advantage against the standard of care [25][26][27].
Among the mechanisms of resistance to mTORC1 inhibitors, feedback activation of PI3K/AKT signaling upon mTORC1 inhibition is of particular therapeutic interest [28]. When only mTORC1 and not mTORC2 is inhibited, AML cells exhibit increased AKT phosphorylation, amplifying survival signals, and activating PI3K [29]. This paradoxical activation contributes to the lack of efficacy of rapamycin and rapalogs and provides a strong rationale for the concomitant inhibition of mTORC1 and mTORC2 and the dual targeting of mTOR and PI3K or AKT [11].
Simultaneous inhibition of mTORC1 and mTORC2 is achieved by ATP-competitive kinase inhibitors like OSI-027, AZD8055 and AZD2014, TAK-228, and PP242 (see Figure 2 for chemical structures), all of which abrogate paradoxical activation of AKT [30].
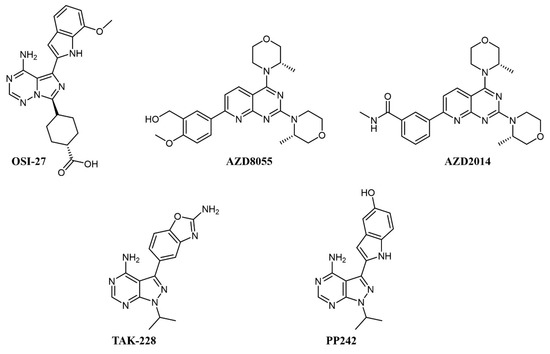
Figure 2. Chemical structure of dual mTOR complex 1 (mTORC1)/mTORC2 inhibitors used for acute myeloid leukemia (AML) treatment.
In preclinical studies on AML cell lines, OSI-027 showed significant inhibition, both as a single agent and in combination with standard chemotherapy, such as cytarabine [31][32]. In a first-in-man, dose-finding clinical trial on solid tumors, OSI-027 inhibited mTORC1/mTORC2 in a dose-dependent manner, but the maximum tolerated dose did not allow significant target engagement in tumor [33]. AZD8055 fully abrogates the PI3K feedback activation and shows remarkable efficacy and no notable toxicity on xenograft models [34]. In clinical trials, AZD8055 showed reasonable toxicity [35] but was later discontinued [36]. Its analog, AZD2014 (vistusertib), had superior pharmacokinetics [36] and a good tolerability profile [37]; on AML cells, it showed synergistic effects with the antibody-drug conjugate gemtuzumab ozogamicin by activation of lysosomal function [38] and with the pan-PIM inhibitor AZD1208 [39].
PP242 also shows better inhibition of mTORC1 and mTORC2 activity and suppression of feedback PI3K/AKT activation than rapamycin and achieved good efficacy with no toxicity in murine models [40]; similar to AZD2014, PP242 showed synergy with gemtuzumab ozogamicin by an analogous mechanism [41]. MLN0128 (formerly TAK-228) is a selective ATP-competitive mTORC1/2 inhibitor that induces apoptosis in cell lines with amplified AKT/mTOR signaling, including AML cell lines, without affecting healthy cells [42]. MLN0128 has entered various clinical trials, including against hematologic malignancies, such as relapsed lymphoma (NCT02727777), showing preliminary therapeutic activity [43].
Several mTOR/PI3K dual inhibitors have been reported, many of which have been tested on AML (Figure 3) [44]; interestingly, cellular studies on primary blasts collected from AML patients evidenced that high PI3K signaling was associated with resistant samples, and dual PI3K/mTOR inactivation was proven to be cytotoxic also to leukemia-initiating cells [45].
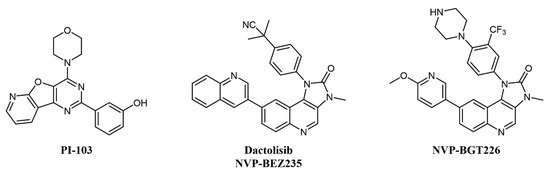
Figure 3. Chemical structure of dual mTOR/PI3K inhibitors used for AML treatment.
Despite the straightforward rationale of concomitant inhibition of PI3K and mTOR, toxicity concerns have been raised over the blocking of PI3K activity, as this kinase is key in a series of important cellular processes [46]. One of the first compounds of this type to be developed was PI-103 (Figure 3), which provided proof-of-concept for the therapeutic potential of double PI3K/mTOR agents on AML cells by inhibiting leukemic proliferation, clonogenicity of leukemic progenitors, and inducing mitochondrial apoptosis [47] but did not enter clinical trials due to limited solubility and extensive metabolism [48]. Other PI3K/mTOR inhibitors assessed on AML are two imidazoquinoline analogs, dactolisib (NVP-BEZ235) and NVP-BGT226 (Figure 3). Dactolisib is endowed with pan-PI3K/mTOR specificity [49]. When tested on 21 primary AML samples and human leukemic cell lines, the compound reduced their proliferation rate, inducing a strong apoptotic response in AML cells without affecting normal CD34+ progenitor cells’ survival [50]. Interestingly, both NVP-BGT226 and dactolisib suppress AKT signaling pathways and show potent antiproliferative effects, but while NVP-BGT226 has potent pro-apoptotic effects in vitro as well as ex vivo on leukemic blasts, dactolisib causes G1/G0 arrest and prevents the induction of apoptosis [51]. Dactolisib entered a Phase I clinical trial on 22 patients with relapsed or refractory acute leukemia (NCT01756118) in 2012, with an overall response rate of 30% and a sustained molecular remission in a single patient [52].
Concomitant dual inhibition of proviral integration site for Moloney murine leukemia virus 1 (PIM1) with other molecular targets has also been proposed as a treatment strategy in AML and evaluated in preclinical studies. PIM proteins are a family of three Ser/Thr kinases responsible for cell cycle regulation, anti-apoptotic activity, and the activation of receptor tyrosine kinases. PIM signaling has a role in defining cell fate, including senescence, cell cycle regulation, apoptosis, metabolism, invasion, and metastasis; PIM1 mRNA levels are increased in acute myeloid leukemia (AML); thus, the combination of a PIM kinase inhibitor with an mTOR inhibitor is expected to offer greater antitumor effects in AML than either inhibitor alone [53]. The combination of dual mTOR inhibitor AZD2014 and pan-PIM inhibitor AZD1208 (Figure 4) effectively reduces protein synthesis by simultaneous inhibition of the mTORC1/2 pathway and induces apoptosis in AML cells [39]. In a similar study, the PIM inhibitor AZD1897 (Figure 4) and the AKT kinase inhibitor AZD5363 (capivasertib, Figure 4) showed synergistic cytotoxicity in AML associated with mTOR and MCL1 pathway repression [54]. It will be interesting to evaluate dual PIM/PI3K inhibitors, such as IBL-202 [55][56] (undisclosed structure) or IBL-302 [57] (Figure 4), in this setting (see also below). SEL24-B489 (MEN1793; Figure 4) is a dual-type I PIM/FLT3 inhibitor based on the benzoimidazole scaffold showing broad activity in AML cell lines and primary AML blasts and efficacy on AML xenografts [58]; SEL24-B489 entered Phase I/II clinical trials on AML in 2017 (NCT03008187). Imidazo-pyridazine SGI-1776 (Figure 4) has a similar selectivity profile and inhibits the growth of both chronic lymphocytic leukemia (CLL) [59] and AML [60] cell lines, but was discontinued after entering clinical trials, including on relapsed/refractory leukemias (NCT01239108), for dose-limiting cardiac toxicity.
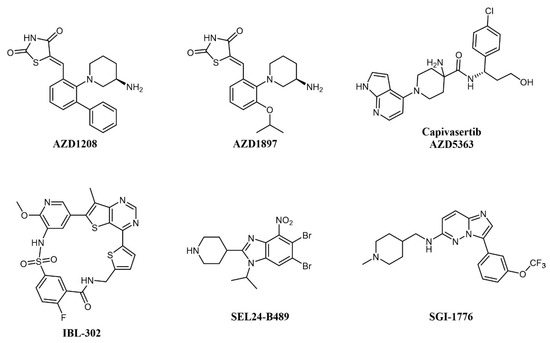
Figure 4. Chemical structure of multi-targeting PIM inhibitors used for AML treatment. The structure of the phosphatidylinositol 3-kinase/protein kinase B (AKT) inhibitor Capivasertib (used in combination with AZD1897) is also reported.
Genetic alterations of FLT3 occur in about 30% of AML cases, more commonly as an internal tandem duplication (FLT3-ITD, 25% of new cases) or as mutations in the tyrosine kinase domain (FLT3-TKD, 7–10% of new cases) [9][61]. In particular, WHO identifies FLT3-ITD as affecting the clinical outcome of AML, conferring poor prognosis, and negatively impacting patient management. [62][63]. As in AML patients, redundant activation of multiple signal transduction pathways, such as PI3K/AKT, MAPK, and JAK/STAT, was observed and linked to poor prognosis early on, the targeting of the oncogenic signal at more than one level in FLT3-targeted therapies has been long recognized as a promising strategy for AML treatment [64]]. Dual inhibition of FLT3 in AML has been recently and exhaustively reviewed ([65] and references within); we will focus here on compounds with specific dual kinase inhibitory profile which have undergone clinical investigations in AML.
Of notice, dual FLT3/JAK2 inhibitor pacritinib (SB1518; Figure 5) [66] strongly inhibits FLT3 auto-phosphorylation and its downstream signaling pathways in AML cell lines and has efficacy in FLT3-ITD driven AML murine xenograft models [67]. Pacritinib has undergone clinical trials in combination for the treatment of AML in FLT-ITD-bearing (NCT02323607) and older patients (NCT02532010). Crenolanib (CP-868–596; Figure 5) is a type I inhibitor of PDGF with sub-nanomolar potency against FLT3 [68]. Crenolanib shows remarkable activity against resistance-conferring mutations of the kinase domain [69] and has entered a number of clinical trials against various indications, including two Phase II studies on relapsed/refractory AML (NCT01657682 and NCT03250338).
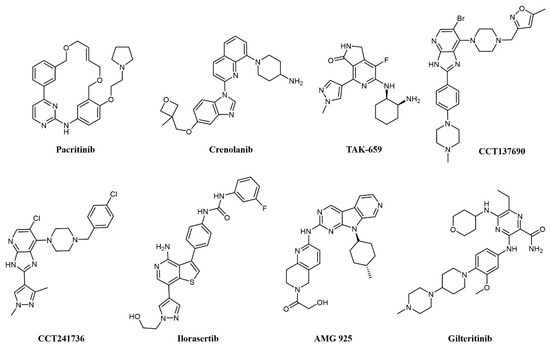
Figure 5. Chemical structure of multi-target fms-like tyrosine kinase 3 (FLT3) inhibitors used for AML treatment.
Spleen tyrosine kinase (SYK) was also recognized early as an important signaling partner of FLT3 [70], and overexpression of SYK confers resistance to selective FLT3 inhibitors [71]. Pyrido indolinone TAK-659 (Figure 5) was developed as an SYK inhibitor and showed strong activity against FLT3 and demonstrated efficacy against AML cell lines and in animal models [72]. TAK-659 has entered clinical trials on a number of indications, including a Phase I/II study as a single agent against relapsed or refractory AML (NCT02323113) to identify the maximum tolerated dose and evaluate preliminary efficacy on the disease.
Aurora kinases are a family of highly conserved serine-threonine protein kinases that have a key role in mitosis; overexpression of Aurora A has been consistently demonstrated in AML cell lines and patient cohorts [73]. Aurora kinase inhibitors were identified as promising agents in the treatment of FLT3-ITD-associated AML [73], providing a rationale for the development of dual FLT3-Aurora kinase inhibitors. The dual Aurora/FLT3 inhibitor CCT137690 (Figure 5) was identified following a medicinal chemistry program starting from an in-house Aurora inhibitor series [74] and was shown to inhibit the growth of FLT3 inhibitor-resistant AML cell lines [73]. CCT241736 (Figure 5) is a more advanced analog of CCT137690, with a favorable selectivity and PK profile and no hERG inhibition [74]. CCT241736 has significant in vivo efficacy against FLT3-ITD human tumor xenograft models and against FLT3-ITD cells with high in vitro relative resistance to the FLT3 inhibitors quizartinib and sorafenib. CCT241736 was also effective on primary samples from AML patients, including those with disease resistance to FLT3 inhibitor quizartinib [75]. Ilorasertib (ABT-348; Figure 5), an Aurora/VEGF inhibitor also potent against FLT3 [76], demonstrated clinical response in 3 out of 38 AML patients in a Phase 1 dose-escalation clinical study [77].
Cyclin-dependent kinase 4 (CDK4) is a downstream effector of growth factor activation, involved in the p16INK4a-CDK4-Rb axis in cancer development, which has a significant role in AML [78]. In preclinical studies, orally available FLT3/CDK4 dual kinase inhibitor AMG-925 (Figure 5) showed efficacy in AML tumor models and inhibited signaling in sorafenib-resistant AML cell lines [78].
In addition, c-Fes, a non-receptor tyrosine kinase involved in hematopoietic cells growth, survival, and differentiation as well as innate immune responses, is implicated in AML as a signaling partner of FLT3 [79]. In a recent work, a series of c-Fes inhibitors with anti-FLT3 activities in the nanomolar range have been shown to cause growth arrest and induce apoptosis in FLT3-ITD+ AML cells, and specific activity to FLT3-resistant cell lines [80].
Another RTK associated with poor prognosis and resistance to therapy in AML is AXL [81][82], a member of the Tyro3-Axl-Mer (TAM) family involved in a number of other cancers [83]. Gilteritinib (ASP2215; Figure 5) is a type I kinase inhibitor based on a pyrazine-carboxamide scaffold showing sub-nanomolar potency against FLT3 and AXL, activity against resistance-conferring mutations of FLT3 in cells, and a favorable pharmacokinetic profile granting activity on AML xenograft models [84]. Gilteritinib entered various clinical trials on AML, including the Phase III multicenter ADMIRAL trial (NCT02421939) comparing gilteritinib to chemotherapy in relapsed/refractory AML with FLT3-mutations [85]. Excitingly, gilteritinib significantly extended the overall survival of the treated patients and showed a higher percentage of patients with remission compared to salvage chemotherapy while showing less adverse events [86]. On the grounds of these results, gilteritinib was approved by the US Food and Drug Administration (FDA) and by the European Commission for the treatment of relapsed or refractory AML.
3. Dual Kinase Targeting in CML
Chronic myelogenous leukemia (CML) is a hematopoietic stem cell disease characterized by the Philadelphia (Ph) chromosome, a shortened chromosome 22 that results from the translocation between chromosomes 9 and 22, that is present in over 90% of CML patients [87]. The fusion gene BCR-ABL (breakpoint cluster region-Abelson leukemia virus) resulting from this translocation encodes the BCR-ABL fusion tyrosine kinase, which causes cell cycle deregulation, apoptosis, and affects DNA repair and differentiation [88][89]. The development of tyrosine kinase inhibitors changed the therapeutic options for CML patients dramatically, improving the 10-year survival rate from approximately 20% to 80–90% [90]. The BCR-ABL inhibitor imatinib was the first targeted therapy approved for the treatment of CML, and the first protein kinase inhibitor approved as a cancer treatment [1][91]. Imatinib quickly became the therapeutic standard for the treatment of CML, owing to the fact that frontline therapy was found to induce durable responses in a high proportion of patients [92]; despite these impressive results, resistance to imatinib treatment emerged as a clinical problem, with a fraction of patients failing to achieve complete hematological response by 3 months (10% of patients) or complete cytogenic response (25% of patients) by 18 months after therapy start [92][93], and a higher rate of resistance among patients with advanced phase CML [94].
Various mechanisms of resistance to tyrosine kinase inhibitor (TKI) treatment in CML have been reported, mainly caused by point mutations of the kinase domain [95], target gene amplification [96], and activation of alternative signaling pathways [97]. Among the latter, the most characterized cooperating pathway involves the avian sarcoma viral oncogene homolog (SRC) Family Kinases (SFKs), whose activation has been shown to induce a BCR-ABL independent mechanism of imatinib resistance [98][99]; furthermore, phosphorylation (activation) of BCR-ABL by SFKs is required for full oncogenic activity [100]. This provides a strong rationale for the use of dual SFK/ABL inhibitors in Ph+ CML.
There are eight structurally related SFKs; the family is involved in RTKs, integrin, GPCRs, and immunoreceptor signaling [101]. Interestingly, the domain organization of ABL and SRC has significant homology [102], making possible the development of dual ATP-competitive SRC-ABL inhibitors. There are now five commercially available tyrosine kinase inhibitors for the treatment of Ph+ CML: imatinib, dasatinib, nilotinib, bosutinib, and ponatinib; of these, dasatinib and bosutinib (Figure 6) are dual SRC-ABL inhibitors [90]. Other advanced dual SRC-ABL inhibitors include FB2, a N-(thiazol-2-yl)pyrimidin-4-amine derivative (structure not completely disclosed) which shows in vitro and in vivo activity against TKI-resistant CML cell lines [103][104], and bafetinib (INNO-406, NS-187; Figure 6), an orally available inhibitor with activity on a number of ABL mutations which also selectively inhibits Lyn over other SRC family members and is able to penetrate the central nervous system (CNS) in murine models [105][106]. In a Phase I clinical trial on CML patients resistant or intolerant to imatinib and second-generation inhibitors, bafetinib achieved a 19% cytogenetic response rate [107]. Dasatinib (BMS-354825; Figure 6) was the first dual SRC-ABL inhibitor to enter the clinic and was developed starting from a series of substituted thiazole-5-carboxamides with activities against SRC and ABL and antiproliferative activity in CML cell lines and xenograft models [108]; besides SRC and ABL, dasatinib binds over 30 kinases, including major regulators of the immune system [109]. Dasatinib was initially approved in 2006 for the treatment of CML and Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) patients resistant to therapy, including imatinib [110]; when compared with imatinib in a Phase III clinical trial at a dose of 100 mg/day, it showed higher molecular response rates [111]. Dasatinib has been the object of more than 300 clinical trials on CML and a number of other pathologies [112]. More recent clinical trials have shown encouraging efficacy of dasatinib at a lower dose, suggesting that future CML treatment could have a better safety profile and lower cost of care [113].
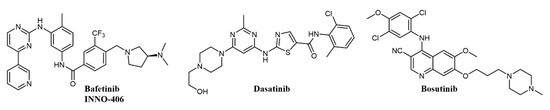
Figure 6. Chemical structure of dual SRC/ABL inhibitors used for chronic myelogenous leukemia (CML) treatment.
Bosutinib (SKI-606, Bosulif; Figure 6), also a dual SRC-ABL inhibitor, binds over 45 kinases with a selectivity profile different from dasatinib [114]. Bosutinib was developed from a series of 4-phenylamino-3-quinolinecarbonitriles SRC inhibitors with activity in vivo [115]. Bosutinib is able to overcome the majority of BCR-ABL mutations conferring resistance to imatinib, with the exception of T315I and V299L [116]. In clinical trials on the treatment of chronic phase CML, bosutinib has shown durable efficacy and manageable toxicity in patients resistant or intolerant to imatinib [117] and achieved higher and faster molecular response than imatinib as a first-line treatment [118]. Accordingly, bosutinib was first approved in 2012 for the treatment of resistant CML [119] and in 2017 for first-line treatment of chronic phase CML [90][120].
4. Dual Kinase Targeting in Lymphoid Tumors
The most recent WHO classification of tumors of hematopoietic and lymphoid tissues (WHO Classification of Tumors, Revised 4th Edition, 2017) reports six classes of lymphoid neoplasms of B-, T-, NK- and dendritic cell origin. Among them, several kinase-addicted diseases can be recognized. Ph+ ALL is a primarily pediatric cancer characterized by the presence of the t(9;22) (q34.1; q11.2) translocation and expression of the fusion oncogene BCR-ABL, akin to Ph+ CML. Hence, similar to CML, the use of imatinib brought great clinical improvement to patients [121]. Unfortunately, in contrast to CML, relapses are frequent due to the development of resistant disease, highlighting the need to develop new multi-targeted agents.
Several studies identified SFKs as key players in the development of Ph+ ALL, where they cooperate with the BCR-ABL fusion kinase to induce transformation [122]. Hence, co-targeting of BCR-ABL and SFKs by the dual ABL/SRC inhibitor dasatinib (Figure 6) allowed long-term survival of mice with Ph+ B-ALL [123]. This aminothiazole compound binds to the active conformations of ABL and SFKs with similar potency and induced complete remission of leukemia in imatinib-resistant patients [124]. Besides Ph+ ALL, other lymphoblastic leukemias can respond to ABL inhibitors, such as T-ALL expressing the NUP214-ABL1 fusion, but again, relapses are frequent. A relevant role of the SRC family kinases, in particular the Leukocyte C-Terminal Src Kinase (LCK), was identified in this disease as well [125]. The involvement of LCK suggested the use of dual ABL/SRC inhibitors. Indeed, NUP214-ABL1-positive cells and xenografts were exquisitely sensitive to dasatinib, and a patient with NUP214-ABL1 fusion was reported to obtain a complete remission with dasatinib [126].
Overexpression of cyclin D1 and consequent activation of CDK4/6 is a hallmark of mantle cell lymphoma (MCL); the CDK4 inhibitor palbociclib has shown modest clinical activity but was found to sensitize MCL cells to PI3K inhibitors [127]. Therefore, concomitant inhibition of CDKs and PI3K was pursued: ON123300 (Figure 7) is a novel pyrido-pyrimidine compound that inhibits CDKs 4 and 6, as well as ARK5 and PI3K-δ. Exploiting dual inhibition of CDK4/Rb and PI3K/AKT/mTOR circuits, the compound showed potent antitumor activity in vitro and in vivo [127]. Taking a similar approach, Natoni and colleagues aimed to simultaneously block two cell cycle regulators, CDC7 and CDK9, in CLL cells [128]. The authors used a pyrrolo-pyrimidinone compound, PHA-767491 (Figure 7), that is able to prevent cell division by blocking CDC7-induced activation of DNA replication origins [129]. PHA-767491 inhibited CLL cell proliferation induced by IL-4, a common mechanism of drug resistance in these cells. Surprisingly, the inhibitor also induced apoptosis in quiescent CLL cells from patients, probably through inhibition of CDK9-dependent transcription of MCL1. Again, this work highlights the value of dual targeting. The same compound was active in multiple myeloma cells [130], as well as in myeloid and solid tumors [131][132]. However, caution must be used because PHA-767491 also suppresses activation of normal T lymphocytes, potentially impairing antitumor immunity [133].
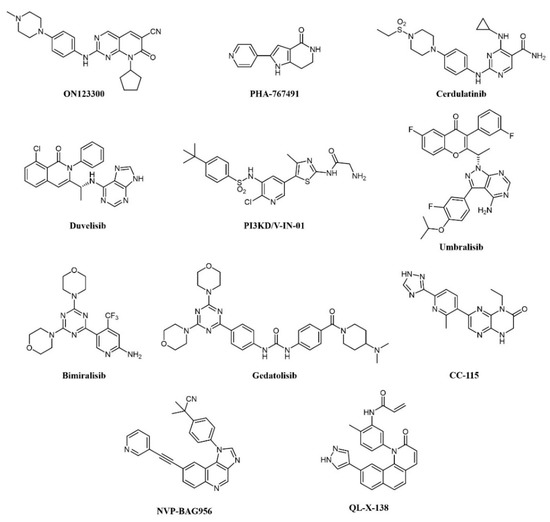
Figure 7. Chemical structure of dual kinase inhibitors used for lymphoid tumors.
More recently, the precise elucidation of other pathways involved in cell transformation led to the design of a dual SYK/JAK kinase inhibitor for the treatment of lymphocytic leukemias and lymphomas. In these diseases, constitutive activation of SYK and JAK kinases has been observed [134][135]. In CLL and B-cell non-Hodgkin’s lymphoma (NHL), SYK is involved both in Bruton’s Tyrosine Kinase (BTK) pathway activation and IL4-driven resistance to BTK inhibitors, while JAK kinases transduce cytokine signaling, which supports tumor growth. A combination of SYK and JAK inhibitors synergistically inhibits cell proliferation, better than any single inhibitor. Hence, a dual SYK/JAK phenylamino-pyrimidine inhibitor, cerdulatinib (PRT062070; Figure 7), induced apoptosis in cancer cells but not in normal B cells, prevented drug resistance, and caused tumor inhibition in mice [136][137][138]. Cerdulatinib inhibits all JAK family kinases and is also efficacious in models of autoimmunity. The compound is currently under clinical investigation for relapsed/refractory CLL and other B-cell malignancies: In a Phase I trial, complete SYK and JAK pathway inhibition was achieved in whole blood of patients at tolerated exposures after oral administration. Objective tumor responses were observed in CLL and follicular lymphoma (FL) patients, including two complete responses. Three of eight CLL patients (38%) achieved a durable partial response lasting >200 days [139][140].
The involvement of class I PI3Ks in the onset of B-cell lymphoma is well documented [141]. First-generation pan-PI3K inhibitors have shown limited clinical efficacy in hematological cancers, probably due to unspecific inhibition of all isoforms and ensuing toxicity. Recently, isoform-selective inhibitors have shown better therapeutic efficacy. Duvelisib (IPI-145, INK-1197; Figure 7) is a dual PI3Kδ/γ quinoline inhibitor that shows selectivity over other class I PI3Ks [142][143]. In a Phase I trial in CLL patients, 56% and 83% responses were observed in chemotherapy-resistant and treatment-naïve patients, respectively. Similar results were obtained in a Phase II study in FL patients and a larger Phase III trial in patients with advanced hematological diseases, including NHL. The compound has now been approved by the FDA for the treatment of CLL and FL patients relapsed or refractory to chemotherapy [144][145]. PI3KD/V-IN-01 is a compound that simultaneously and selectively inhibits PI3Kδ and PIK3C3/Vps34, a class III PI3K isoform that can induce cytoprotective autophagy [146]. The compound showed superior antiproliferative activity against AML, CLL, and Burkitt’s lymphoma primary cells, compared to a PI3Kδ-only inhibitor (idelalisib) and inhibited tumor growth in vivo. Another PI3Kδ inhibitor, umbralisib (TGR-1202; Figure 7), also blocks casein kinase-1ε (CK1ε). Targeting of CK1ε has been shown to cooperate with idelalisib in hematological malignancies and to alleviate some immune-mediated adverse effects observed with pan-PI3K inhibitors. Thus, umbralisib was shown to possess comparable antitumor activity but lower toxicity in murine models of CLL [147]. The drug is being evaluated in CLL and B-cell NHL patients with encouraging results [148][149].
A number of PI3K inhibitors are described as dual PI3K/mTOR inhibitors. As discussed above, these compounds are likely to achieve a deeper suppression of the entire PI3K/AKT/mTOR pathway, compared to PI3K-specific inhibitors and, indeed, dual PI3K/mTOR inhibition is an established strategy for the treatment of AML. There is also strong evidence that simultaneous PI3K/mTOR inhibition induces better preclinical antitumor activity than single targeting in lymphoma [150]. Several PI3K/mTOR dual inhibitors have been developed in recent years, also thanks to a good structural similarity between the two kinases [151]. One such compound, bimiralisib (PQR309; Figure 7), was shown to inhibit all class I PI3K catalytic enzymes and mTOR with low nanomolar potency selectively while sparing the rest of the kinome [152]. Bimiralisib showed antitumor activity in preclinical lymphoma models [150] and is currently in clinical development for lymphoid malignancies. In a Phase I/II trial, clinical benefit was obtained in 8/11 lymphoma patients, including one complete remission (CR) [153]. Expanded cohorts are currently being enrolled in the trial. Gedatolisib (WYE-129587/PKI-587/PF-05212384; Figure 7), a PI3Kα/mTOR inhibitor, has been investigated in preclinical leukemia models, including T-ALL and Ph-like ALL, with very good results [154][155]. Dactolisib (see also above; Figure 3) inhibits cell growth and induces apoptosis in ALL and follicular lymphoma [156][157][158][159][160]. However, clinical investigations have mainly focused on solid tumors. Compound PI-103 (Figure 3) was tested on a panel of T-ALL cell lines and primary patients’ cells with constitutive activation of PI3K/Akt/mTOR pathway, showing potent cytotoxicity accompanied by complete inactivation of AKT, p70S6K, ribosomal S6 protein, and 4E-BP1 [161]. The importance of the mTOR pathway in cancer development further supported other bi-specific drug development programs, such as the dual mTOR/DNA-PK inhibitor, CC-115, or the PI3K/PDK1 inhibitor, NVP-BAG956 (Figure 7). DNA-PK is a Ser/Thr kinase that phosphorylates AKT in response to DNA damage, and, therefore, its inhibition can enhance the efficacy of anticancer treatments, and, in particular, of AKT/mTOR inhibitors. Indeed, CC-115 showed good preclinical and clinical activity in CLL [162][163]. NVP-BAG956, acting at two levels along the PI3K/mTOR pathway, showed potent cytotoxic effects against T-ALL cell lines and primary patients’ samples, outperforming drugs hitting a single target, such as PI3K, mTOR, or AKT inhibitors, which achieved comparable effects when combined, however [164].
In B-cell neoplasms, the roles of BTK and PI3K have been well established. Indeed, inhibitors of these kinases, such as ibrutinib and idelalisib, have shown efficacy against these cancers [165]. Liu et al. from the ShanghaiTech University (China) designed a series of dual BTK/PI3Kdelta pyridinone inhibitors, which simultaneously block BTK and PI3K pathways and inhibit Burkitt’s lymphoma cells growth [166][167]. In CLL cells, the dual PI3K/PIM kinase inhibitor IBL-202 suppressed cell proliferation and migration in hypoxic conditions where idelalisib failed [56]; the compound also showed efficacy in multiple myeloma [55]. Along similar lines of research, a BTK/MNK (Mitogen-Activated Protein Kinase Interacting Kinase) dual inhibitor was developed for lymphoma and leukemia (QL-X-138; Figure 7), which interestingly establishes covalent binding to BTK and a standard reversible binding to MNK [168]. MNK kinases are downstream effectors of various intracellular pathways and can induce transformation: their inhibition synergizes with upstream kinases blockade. QL-X-138 showed superior activity compared to a BTK inhibitor.
5. Other Dual Activity Inhibitors in Leukemia
As cancer is generally a multifactorial disease, effective therapeutic approaches make use of a combination of drugs endowed with different mechanisms of action (e.g., a kinase inhibitor in association with an antimetabolite [169]). While dual kinase inhibitors hit different targets belonging to the same protein family, a new concept of “dual-targeting” has been developed in recent years that makes use of a single drug endowed with more than one mechanism of action. Such compounds are obtained by merging the pharmacophore moieties of two different drugs into a single compound. Besides blocking different pathways at the same time, these compounds also have the advantage of single and more predictable pharmacokinetics. As an example, several reports have shown the synergistic potential of kinase and HDAC inhibitor combinations in cancer [170][171][172]. Hence, merging the two activities in one molecule would be a further step to increasing therapeutic activity. A medicinal chemistry effort aimed at incorporating HDAC inhibitory function into a PI3K inhibitor pharmacophore led to the identification of fimepinostat (CUDC-907; Figure 8), a PI3K/HDAC dual inhibitor for the treatment of lymphoid cancers [173][174][175]. In a Phase I trial, the drug achieved an objective response in 5/9 patients with relapsed or refractory Diffure Large B-Cell Lymphoma (DLBCL) [176]. While its safety profile was consistent with those of FDA-approved HDAC inhibitors and PI3K inhibitors, fimepinostat showed superior tolerability as it did not lead to the severe side effects associated with HDAC and PI3K inhibitors (e.g., hepato- and cardiotoxicities) [176]. Notably, in animal models, the co-administration of a PI3K inhibitor and an HDAC inhibitor was not tolerated, whereas the use of fimepinostat did not show signs of appreciable toxicity at therapeutic doses [177]. Preclinical studies also suggested a potential application in the treatment of CLL [178], mantle cell lymphoma (MCL) (174), and AML [179]. The compound is currently undergoing Phase 1 clinical investigation in children and young adults with relapsed or refractory solid tumors, CNS tumors, or lymphoma (ClinicalTrials.gov Identifier: NCT02909777).
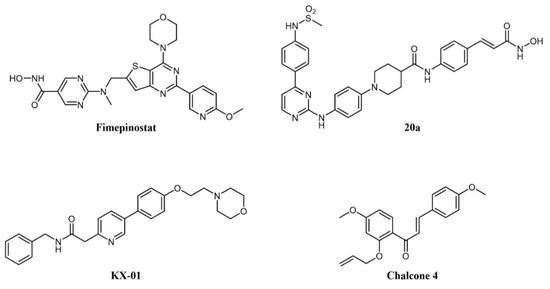
Figure 8. Chemical structure of dual-target kinase inhibitors.
Several rationally designed JAK2/HDAC6 inhibitors have also been reported [180]. The most promising compound in the series (20a in the paper; Figure 8) showed comparable potency against the targets (IC50 JAK2 = 8 nM; IC50 HDAC6 = 46 nM), which prolonged the survival of mice in an AML xenograft model, and counteracted spleen enlargement. A preliminary evaluation of the pharmacokinetic profile was also performed.
Tirbanibulin (KX-01; Figure 8) is a novel compound equipped with tyrosine kinase inhibitory as well as antimicrotubule activity. It is, in fact, a peptidomimetic compound that inhibits at the same time SRC kinase (through interference with substrate binding) and microtubule polymerization, thus providing a comprehensive suppression of the cell migration machinery [181]. The compound is under clinical investigation for several indications, including lymphoma (ClinicalTrials.gov Identifier: NCT00646139). A recent report identified natural chalcone compounds as dual FLT3 and microtubule polymerization inhibitors [182]. The authors then derived synthetic analogs, the most active of which (chalcone 4; Figure 8) simultaneously reversed activation of drug-resistant FLT3-ITDD835Y mutant and tubulin polymerization in vitro.
6. PROTACs
One of the most interesting recent developments in this field relates to Proteolysis Targeting Chimeras (PROTACs) compounds. It is a new class of bifunctional molecules, composed of 1) a ligand that binds a biomolecular target; 2) a ligand that recognizes the E3 ubiquitin ligase; 3) a linker between 1 and 2 [183][184][185][186][187]. The interaction with E3 ubiquitin ligase promotes the ubiquitination of the biomolecular target and, consequently, the degradation of the protein by the proteasome. By this mechanism, PROTACs can reduce the intracellular concentration of a given protein, leading to a prolonged and marked inhibitory effect. Despite the very recent development of such a strategy, several papers and reviews on the PROTACs strategy can be found in the literature [188][189][190][191]. A general scheme of the PROTACs strategy is reported in Figure 9.
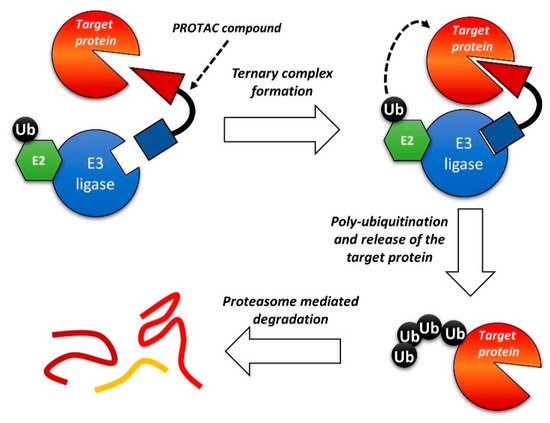
Figure 9. Schematic representation of the Proteolysis Targeting Chimeras (PROTAC) strategy.
The PROTAC approach was recently applied to BCR-ABL-driven leukemia. Various reports identified cyclin-dependent kinase 8 (CDK8) as a key mediator of Ph+ B-ALL, also through kinase-independent functions. In this context, CDK8 was shown to activate the PI3K/mTOR pathway. Thus, combined inhibition of CDK8 and mTOR was pursued: The authors showed the efficacy of a dual CDK8/mTOR inhibitor, YKL-06-101 (Figure 10), which is also a CDK8 degrader [192]. Degradation of CDK8 was necessary because the simple biochemical inhibition was insufficient to cause cell death due to the kinase-independent activity of CDK8. The compound was derived from the optimization of Torin1, a potent mTOR inhibitor[193], towards dual mTOR/CDK8 inhibition; this fragment was then linked to thalidomide to add the degrader function. YKL-06-101 was equally active in Ph+ and Ph- B-ALL cells. Interestingly, this compound recapitulates both the dual kinase and the PROTAC approach.
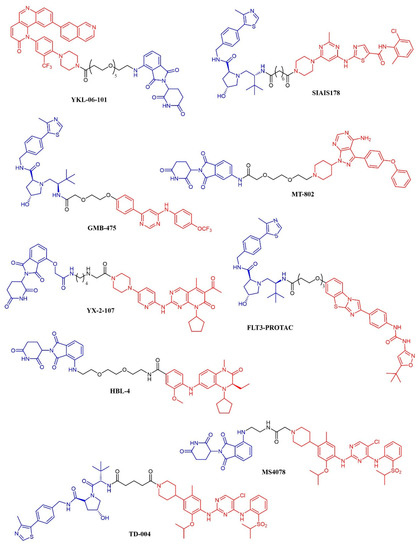
Figure 10. Chemical structure of kinase inhibitor derived PROTACS. Ubiquitine ligase recognizing moiety is depicted in blue; kinase recognizing moiety is depicted in red; linker is depicted in black.
A series of dasatinib-based PROTACs has been recently reported (194). The compounds differed from the linker length and were preliminarily screened against K562 cells. The most promising compound (SIAIS178; Figure 10) was found to induce the selective degradation of BCR/ABL kinase and several mutants thereof. Notably, the modification of the dasatinib structure caused a narrower selectivity profile of the PROTAC compound. When tested in a murine xenograft model of K562 cells, the compound effectively reduced the tumor progression but did not outperform dasatinib. The GNF-5 allosteric modulator of BCR-ABL kinase has been modified according to the PROTAC strategy, leading to GMB-475 (Figure 10). The compound induced a time- and dose-dependent degradation of the target protein. The potential antineoplastic activity was assessed in K562 and Ba/F3 cell lines and primary CML patient samples. GMB-475 induced the degradation of the target protein in CML stem cell populations also, but without causing marked apoptosis, further confirming that CML stem cells’ survival is not dependent on BCR-ABL (195).
MT-802 (Figure 10) is an ibrutinib-based PROTAC active against primary CLL patient samples. The compound outperformed the parent kinase inhibitor, resulting in the ability to induce the degradation of both wt-BTK and the ibrutinib-resistant mutant BTKC481S {196}. No in vivo experiments have been reported so far.
YX-2-107 (Figure 10) is a palbociclib-derived PROTAC that induces the degradation of CDK6 in Ph+ ALL cells [194]. When tested in vivo in Ph+ ALL xenograft models, the compound suppressed the number of cells in the S phase with the same potency as palbociclib. However, the PROTAC derivative abrogated the expression of CDK6 (that is, instead, upregulated upon treatment with palbociclib) and, at a lower extent, of CDK4. The pharmacokinetic properties of YX-2-107 were not optimal, and the compound needs further improvement.
The enhancement in the selectivity of PROTAC derivative of kinase inhibitor was also observed for a quizartinib-based PROTAC, that resulted active both in vitro and in vivo against AML models [195]. The compound (named FLT-PROTAC; Figure 10) was able to abrogate the FLT3-ITD expression and showed higher efficacy than quizartinib in vitro and an MV4-11 xenograft model.
HBL-4 (Figure 10) is a dual BRD4/PLK1 degrader, obtained through the modification of the dual inhibitor BI2536 according to the PROTAC strategy [196]. HBL-4 effectively reduced the tumor growth in an MV4-11 xenograft AML model and outperformed the parent compound both in vitro and in vivo.
Finally, the same approach was recently developed in the context of ALK-positive lymphoma: three groups described degraders of ALK kinase based on phenylamino-pyrimidine ALK inhibitors (NVP-TAE684 or ceritinib) and either pomalidomide (compounds 9, 10, 11, 12 [197]; compounds MS4077 and MS4078 [198]) or a different von Hippel-Lindau ligand (TD-004 [199]). The latter was shown to have in vivo efficacy in a xenograft model.
References
- Le Coutre, P.; Mologni, L.; Cleris, L.; Marchesi, E.; Buchdunger, E.; Giardini, R.; Formelli, F.; Gambacorti‐Passerini, C. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J. Natl. Cancer Inst. 1999, 91, 163–168.
- Hochhaus, A.; Druker, B.; Sawyers, C.; Guilhot, F.; Schiffer, C.A.; Cortes, J.; Niederwieser, D.W.; Gambacorti-Passerini, C.; Stone, R.M.; Goldman, J.; et al. Favorable long-term follow-up results over 6 years for response, survival, and safety with imatinib mesylate therapy in chronic-phase chronic myeloid leukemia after failure of interferon-alpha treatment. Blood 2008, 111, 1039–1043.
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Ac-cumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550.
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 6219–6226.
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897.
- Mologni, L.; Dekhil, H.; Ceccon, M.; Purgante, S.; Lan, C.; Cleris, L.; Magistroni, V.; Formelli, F.; Gambacorti-Passerini, C. Colorectal Tumors Are Effectively Eradicated by Combined Inhibition of {beta}-Catenin, KRAS, and the Oncogenic Tran-scription Factor ITF2. Cancer Res. 2010, 70, 7253–7263.
- Janne, P.A.; Shaw, A.T.; Camidge, D.R.; Giaccone, G.; Shreeve, S.M.; Tang, Y.; Goldberg, Z.; Martini, J.-F.; Xu, H.; James, L.P.; et al. Combined Pan-HER and ALK/ROS1/MET Inhibition with Dacomitinib and Crizotinib in Advanced Non-Small Cell Lung Cancer: Results of a Phase I Study. J. Thorac. Oncol. 2016, 11, 737–747.
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152.
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and ev-idence. Leukemia 2019, 33, 299–312.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018, 32, 235–248.
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474.
- National Institutes of Health. SEER database. Available online: https://seer.cancer.gov/statfacts/ (accessed on24 November 2020).
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453.
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464.
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731.
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868.
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103.
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol. Life Sci. 2015, 72, 2337–2347.
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic tar-geting. Nat. Rev. Cancer. 2015, 15, 7–24.
- Gentzler, R.D.; Altman, J.K.; Platanias, L.C. An overview of the mTOR pathway as a target in cancer therapy. Expert Opin. Ther. Targets. 2012, 16, 481–489.
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129.
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058.
- Récher, C.; Dos Santos, C.; Demur, C.; Payrastre, B. mTOR, A New Therapeutic Target in Acute Myeloid Leukemia. Cell Cy-cle 2005, 4, 1540–1549.
- Park, S.; Chapuis, N.; Saint Marcoux, F.; Recher, C.; Prebet, T.; Chevallier, P.; Cahn, J.-Y.; Leguay, T.; Bories, P.; et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first re-lapse. Leukemia 2013, 27, 1479–1486.
- Perl, A.E.; Kasner, M.T.; Tsai, D.E.; Vogl, D.T.; Loren, A.W.; Schuster, S.J.; Porter, D.L.; Stadtmauer, E.A.; Goldstein, S.C.; Frey, N.V.; et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in re-lapsed and refractory acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 6732–6739.
- Amadori, S.; Stasi, R.; Martelli, A.M.; Venditti, A.; Meloni, G.; Pane, F.; Martinelli, G.; Lunghi, M.; Pagano, L.; Cilloni, D.; et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: Results of a phase II GIMEMA study (AML-1107). Br. J. Haematol. 2012, 156, 205–212.
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combina-tions and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50.
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382.
- Grzybowska-Izydorczyk, O.; Smolewski, P. MTOR kinase inhibitors as a treatment strategy in hematological malignancies. Future Med. Chem. 2012, 4, 487–504.
- Altman, J.K.; Sassano, A.; Kaur, S.; Glaser, H.; Kroczynska, B.; Redig, A.J.; Russo, S.; Barr, S.; Platanias, L.C. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on Acute Myeloid Leukemia (AML) progenitors. Clin. Can-cer Res. 2011, 17, 4378–4388.
- Eyre, T.A.; Collins, G.P.; Goldstone, A.H.; Cwynarski, K. Time now to TORC the TORC? New developments in mTOR pathway inhibition in lymphoid malignancies. Br. J. Haematol. 2014, 166, 336–351.
- Mateo, J.; Olmos, D.; Dumez, H.; Poondru, S.; Samberg, N.L.; Barr, S.; Van Tornout, J.M.; Jie, F.; Sandhu, S.; Tan, D.S.; et al. A first in man, dose-finding study of the mTORC1/mTORC2 inhibitor OSI-027 in patients with advanced solid malignancies. Br. J. Cancer. 2016, 114, 889–896.
- Willems, L.; Chapuis, N.; Puissant, A.; MacIel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202.
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.L.; Taylor, R.H.; Kaye, S.B.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer. 2012, 107, 1093–1099.
- Pike, K.G.; Malagu, K.; Hummersone, M.G.; Menear, K.A.; Duggan, H.M.E.; Gomez, S.; Martin, N.M.B.; Ruston, L.; Pass, S.L.; Pass, M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: The discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett. 2013, 23, 1212–1216.
- Banerji, U.; Dean, E.J.; Gonzalez, M.; Greystoke, A.P.; Basu, B.; Krebs, M.; Puglisi, M.; Grinsted, L.; Oelmann, E.; Burke, W.; et al. First-in-human phase I trial of the dual mTORC1 and mTORC2 inhibitor AZD2014 in solid tumors. J. Clin. Oncol. 2012, 30 (Suppl. 15), 3004.
- Mizutani, Y.; Inase, A.; Maimaitili, Y.; Miyata, Y.; Kitao, A.; Matsumoto, H.; Kawaguchi, K.; Higashime, A.; Goto, H.; Kurata, K.; et al. An mTORC1/2 dual inhibitor, AZD2014, acts as a lysosomal function activator and enhances gemtuzumab ozogamicin-induced apoptosis in primary human leukemia cells. Int. J. Hematol. 2019, 110, 490–499.
- Harada, M.; Harada, M.; Yamamoto, S.; Tabe, Y.; Benito, J.; Ramirez, S.; Jacamo, R.; Platanias, L.; Matsushita, H.; Fujimura, T.; et al. The novel combination of dual mTOR inhibitor AZD2014 and pan-PIM inhibitor AZD1208 inhibits growth in acute myeloid leukemia via HSF pathway suppression. Oncotarget 2015, 6, 37930–37947.
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.H.; Kornblau, S.M.; Baggerly, K.A.; Jessen, K.; Liu, Y.; Kantarjian, H.; Rommel, C.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689.
- Maimaitili, Y.; Inase, A.; Miyata, Y.; Kitao, A.; Mizutani, Y.; Kakiuchi, S.; Shimono, Y.; Saito, Y.; Sonoki, T.; Minami, H.; et al. An mTORC1/2 kinase inhibitor enhances the cytotoxicity of gemtuzumab ozogamicin by activation of lysosomal function. Leuk. Res. 2018, 74, 68–74.
- Zeng, Z.; Wang, R.Y.; Qiu, Y.H.; Mak, D.H.; Coombes, K.; Yoo, S.Y.; Zhang, Q.; Jessen, K.; Liu, Y.; Rommel, C.; et al. MLN0128, a novel mTOR kinase inhibitor, disrupts survival signaling and triggers apoptosis in AML and AML stem/progenitor cells. Oncotarget 2016, 7, 55083–55097.
- Ghobrial, I.M.; Siegel, D.S.; Vij, R.; Berdeja, J.G.; Richardson, P.G.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Wolf, J.L. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: A phase I dose escalation study in patients with re-lapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am. J. Hematol. 2016, 91, 400–405.
- Zhang, Y.J.; Duan, Y.; Zheng, X.F.S. Targeting the mTOR kinase domain: The second generation of mTOR inhibitors. Drug Discov. Today 2011, 16, 325–331.
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9.
- Pallet, N.; Legendre, C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2013, 12, 177–186.
- Park, S.; Chapuis, N.; Bardet, V.; Tamburini, J.; Gallay, N.; Willems, L.; Knight, Z.A.; Shokat, K.M.; Azar, N.; Viguié, F.; et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008, 22, 1698–1706.
- Raynaud, F.I.; Eccles, S.A.; Patel, S.; Alix, S.; Box, G.; Chuckowree, I.; Folkes, A.; Gowan, S.; Brandon, A.D.H.; Di Stefano, F.; et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: From PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol. Cancer Ther. 2009, 8, 1725–1738.
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030.
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leuke-mia. Clin. Cancer Res. 2010, 16, 5424–5435.
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Rasp, K.H.; Illing, B.; Döhner, H.; Döhner, K.; Schittenhelm, M.M. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol. Cancer. 2013, 12, 1–18.
- Wunderle, L.; Badura, S.; Lang, F.; Wolf, A.; Schleyer, E.; Serve, H.; Goekbuget, N.; Pfeifer, H.; Bug, G.; Ottmann, O.G. Safety and Efficacy of BEZ235, a Dual PI3-Kinase/mTOR Inhibitor, In Adult Patients With Relapsed Or Refractory Acute Leukemia: Results Of a Phase I Study. Blood 2013, 122, 2675.
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Phar-macol. 2013, 85, 629–643.
- Meja, K.; Stengel, C.; Sellar, R.; Huszar, D.; Davies, B.R.; Gale, R.E.; Linch, D.C.; Khwaja, A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br. J. Haematol. 2014, 167, 69–79.
- Reidy, M.; VanDijk, M.; O’Neill, M.; O’Dwyer, M. The Dual PIM/PI3-K Inhibitor Ibl-202 Overcomes Microenvironmental Mediated Resistance in Multiple Myeloma and Prevents PIM1 Induced CXCR4 Upregulation. Blood 2015, 126, 5350.
- Crassini, K.; Shen, Y.; O’Dwyer, M.; O’Neill, M.; Christopherson, R.; Mulligan, S.; Best, O.G. The dual inhibitor of the phos-phoinositol-3 and PIM kinases, IBL-202, is effective against chronic lymphocytic leukaemia cells under conditions that mimic the hypoxic tumour microenvironment. Br. J. Haematol. 2018, 182, 654–669.
- Kennedy, S.P.; O’Neill, M.; Cunningham, D.; Morris, P.G.; Toomey, S.; Blanco-Aparicio, C.; Martínez, S.; Pastor, J.; Eustace, A.J.; Hennessy, B.T. Preclinical evaluation of a novel triple-acting PIM/PI3K/mTOR inhibitor, IBL-302, in breast cancer. On-cogene 2020, 39, 3028–3040.
- Czardybon, W.; Windak, R.; Gołas, A.; Gałezowski, M.; Sabiniarz, A.; Dolata, I.; Salwińska, M.; Guzik, P.; Zawadzka, M.; Gabor-Worwa, E.; et al. A novel, dual pan-PIM/FLT3 inhibitor SEL24 exhibits broad therapeutic potential in acute myeloid leukemia. Oncotarget 2018, 9, 16917–16931.
- Chen, L.S.; Redkar, S.; Bearss, D.; Wierda, W.G.; Gandhi, V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2009, 114, 4150–4157.
- Chen, L.S.; Redkar, S.; Taverna, P.; Cortes, J.E.; Gandhi, V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood 2011, 118, 693–702.
- Nagel, G.; Weber, D.; Fromm, E.; Erhardt, S.; Lübbert, M.; Fiedler, W. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann. Hematol. 2017, 96, 1993–2003.
- Grimwade, D.; Mrózek, K. Diagnostic and Prognostic Value of Cytogenetics in Acute Myeloid Leukemia. Hematol. Oncol. Clin. N. Am. 2011, 25, 1135–1161.
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510.
- Kornblau, S.M.; Womble, M.; Yi, H.Q.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous acti-vation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365.
- Yuan, T.; Qi, B.; Jiang, Z.; Dong, W.; Zhong, L.; Bai, L.; Tong, R.; Yu, J.; Shi, J. Dual FLT3 inhibitors: Against the drug re-sistance of acute myeloid leukemia in recent decade. Eur. J. Med. Chem. 2019, 178, 468–483.
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Hu, C.Y.; Hentze, H.; Tan, Y.C.; Madan, B.; Amalini, C.; Loh, Y.K.; Ong, L.C.; et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–1759.
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Tan, Y.C.; Madan, B.; Amalini, C.; Ong, L.C.; Kheng, B.; Cheong, A.; Zhou, J.; et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011, 1, 1–9.
- Heinrich, M.C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. Crenolanib in-hibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 4375–4384.
- Smith, C.C.; Lasater, E.A.; Lin, K.C.; Wang, Q.; Quino McCreery, M.; Stewart, W.K.; Damon, L.E.; Perl, A.E.; Jeschke, G.R.; Sugita, M.; e t al. Crenolanib is a selective type i pan-FLT3 inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 5319–5324.
- Turner, M.; Joseph Mee, P.; Costello, P.S.; Williams, O.; Price, A.A.; Duddy, L.P.; Furlong, M.T.; Geahlen, R.L.; Tybulewicz, V.L.J. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 1995, 378, 298–302.
- Weisberg, E.L.; Puissant, A.; Stone, R.; Sattler, M.; Buhrlage, S.J.; Yang, J.; Manley, P.W.; Meng, C.; Buonopane, M.; Daley, J.F.; et al. Characterization of midostaurin as a dual inhibitor of FLT3 and SYK and potentiation of FLT3 inhibition against FLT3-ITD-driven leukemia harboring activated SYK kinase. Oncotarget 2017, 8, 52026–52044.
- Lam, B.; Arikawa, Y.; Cramlett, J.; Dong, Q.; De Jong, R.; Feher, V.; Grimshaw, C.E.; Farrell, P.J.; Hoffman, I.D.; Jennings, A. et al. Discovery of TAK-659 an orally available investigational inhibitor of Spleen Tyrosine Kinase (SYK). Bioorg. Med. Chem. Lett. 2016, 26, 5947–5950.
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; e t al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470.
- Bavetsias, V.; Crumpler, S.; Sun, C.; Avery, S.; Atrash, B.; Faisal, A.; Moore, A.S.; Kosmopoulou, M.; Brown, N.; Sheldrake, P.W.; et al. Optimization of Imidazo[4,5-b]pyridine-Based Kinase Inhibitors: Identification of a Dual FLT3/Aurora Kinase In-hibitor as an Orally Bioavailable Preclinical Development Candidate for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 2012, 55, 8721–8734.
- Moore, A.S.; Faisal, A.; Mak, G.W.Y.; Miraki-Moud, F.; Bavetsias, V.; Valenti, M.; Box, G.; Hallsworth, A.; Brandon, A.D.H.; Xavier, C.P.R.; et al. Quizartinib-resistant FLT3-ITD acute myeloid leukemia cells are sensitive to the FLT3-Aurora kinase in-hibitor CCT241736. Blood Adv. 2020, 4, 1478–1491.
- Maitland, M.L.; Piha-Paul, S.; Falchook, G.; Kurzrock, R.; Nguyen, L.; Janisch, L.; Karovic, S.; McKee, M.; Hoening, E.; Wong, S.; et al. Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348) in a phase 1 dose-escalation trial. Br. J. Cancer. 2018, 118, 1042–1050.
- Garcia-Manero, G.; Tibes, R.; Kadia, T.; Kantarjian, H.; Arellano, M.; Knight, E.A.; Xiong, H.; Qin, Q.; Munasinghe, W.; Rob-erts-Rapp, L.; et al. Phase 1 dose escalation trial of ilorasertib, a dual Aurora/VEGF receptor kinase inhibitor, in patients with hematologic malignancies. Investig. New Drugs. 2015, 33, 870–880.
- Keegan, K.; Li, C.; Li, Z.; Ma, J.; Ragains, M.; Coberly, S.; Hollenback, D.; Eksterowicz, J.; Liang, L.; Weidner, M.; et al. Pre-clinical Evaluation of AMG 925, a FLT3/CDK4 Dual Kinase Inhibitor for Treating Acute Myeloid Leukemia. Mol. Cancer Ther. 2014, 13, 880–889.
- Voisset, E.; Lopez, S.; Chaix, A.; Georges, C.; Hanssens, K.; Prébet, T.; Dubreuil, P.; De Sepulveda, P.; FES kinases are re-quired for oncogenic FLT3 signaling. Leukemia 2010, 24, 721–728.
- Weir, M.C.; Hellwig, S.; Tan, L.; Liu, Y.; Gray, N.S.; Smithgall, T.E. Dual inhibition of fes and flt3 tyrosine kinases potently inhibits flt3-itd+ aml cell growth. PLoS ONE 2017, 12, e0181178.
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.-M.; Chuang, S.-E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324.
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leu-kemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452.
- Linger, R.M.A.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Adv. Cancer Res. 2008, 100, 35–83.
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. New Drugs 2017, 35, 556–565.
- Gorcea, C.M.; Burthem, J.; Tholouli, E. ASP2215 in the treatment of relapsed/refractory acute myeloid leukemia with FLT3 mutation: Background and design of the ADMIRAL trial. Future Oncol. 2018, 14, 1995–2004.
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740.
- Sawyers, C.L. Chronic Myeloid Leukemia. N. Engl. J. Med. 1999, 340, 1330–1340.
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. the Biology of Acute Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 164–172.
- Penserga, E.T.P.; Skorski, T. Fusion tyrosine kinases: A result and cause of genomic instability. Oncogene 2007, 26, 11–20.
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709.
- Druker, B.J. Imatinib as a paradigm of targeted therapies. Adv. Cancer Res. 2004, 91, 1–30.
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Gold-man, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417.
- Baccarani, M.; Cortes, J.; Pane, F.; Niederwieser, D.; Saglio, G.; Apperley, J.; Cervantes, F.; Deininger, M.; Gratwohl, A.; Guilhot, F.; et al. Chronic Myeloid Leukemia: An Update of Concepts and Management Recommendations of European Leu-kemia. Net. J. Clin. Oncol. 2009, 27, 6041–6051.
- Bixby, D.; Talpaz, M. Seeking the causes and solutions to imatinib-resistance in chronic myeloid leukemia. Leukemia 2011, 25, 7–22.
- Hare, T.O.; Eide, C.A.; Deininger, M.W.N. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249.
- Le Coutre, P.; Tassi, E.; Varella-Garcia, M.; Barni, R.; Mologni, L.; Cabrita, G.; Marchesi, E.; Supino, R.; Gambacorti-Passerini, C. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 2000, 95, 1758–1766.
- Barouch-bentov, R.; Sauer, K. Mechanisms of drug resistance in kinases. Expert Opin. Investig. Drugs. 2011, 20, 153–208.
- Ptasznik, A.; Nakata, Y.; Kalota, A.; Emerson, S.G.; Gewirtz, A.M. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat. Med. 2004, 10, 1187–1189.
- Donato, N.J.; Wu, J.Y.; Stapley, J.; Gallick, G.; Lin, H.; Arlinghaus, R.; Talpaz, M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003, 101, 690–698.
- Meyn, M.A., 3rd; Wilson, M.B.; Abdi, F.A.; Fahey, N.; Schiavone, A.P.; Wu, J.; Hochrein, J.M.; Engen, J.R.; Smithgall, T.E. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J. Biol. Chem. 2006, 281, 30907–30916.
- Benati, D.; Baldari, C.T. Src Family Kinases as Potential Therapeutic Targets for Malignancies and Immu- nological Disorders. Curr. Med. Chem. 2008, 15, 1154–1165.
- Panjarian, S.; Iacob, R.E.; Chen, S.; Engen, J.R.; Smithgall, T.E. Structure and Dynamic Regulation of Abl Kinases. J. Biol. Chem. 2013, 288, 5443–5450.
- Liu, H.; Li, H.; Feng, Z.; Tai, J.; Meng, Y.; Wang, H.; Xin, H.; Zhang, S., Zuo, M.; Zhang, Y.; et al. Activity of FB2, a novel dual Abl/Src tyrosine kinase inhibitor, against imatinib-resistant chronic myeloid leukemia in vivo and in vitro. Leuk. Lym-phoma 2009, 50, 437–446.
- Yuan, X.; Zhang, Y.; Zhang, H.; Jin, J.; Li, X.; Liu, H.; Feng, Z.; Chen, X. Activity of the potent dual Abl/Src tyrosine kinase inhibitor FB2 against Bcr-Abl positive cell lines in vitro and in vivo. Leuk. Res. 2011, 35, 237–242.
- Kimura, S.; Naito, H.; Segawa, H.; Kuroda, J.; Yuasa, T.; Sato, K.; Yokota, A.; Kamitsuji, Y.; Kawata, E.; Ashihara, E.; et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood 2005, 106, 3948–3954.
- Yokota, A.; Kimura, S.; Masuda, S.; Ashihara, E.; Kuroda, J.; Sato, K.; Kamitsuji, Y.; Kawata, E.; Deguchi, Y.; Urasaki, Y.; et al. INNO-406, a novel BCR-ABL/Lyn dual tyrosine kinase inhibitor, suppresses the growth of Ph+ leukemia cells in the cen-tral nervous system, and cyclosporine A augments its in vivo activity. Blood 2007, 109, 306–314.
- Kantarjian, H.; Le Coutre, P.; Cortes, J.; Pinilla-Ibarz, J.; Nagler, A.; Hochhaus, A.; Kimura, S.; Ottmann, O.G. Phase 1 study of INNO-406, a dual Abl/Lyn kinase inhibitor, in philadelphia chromosome-positive leukemias after imatinib resistance or intolerance. Cancer 2010, 116, 2665–2672.
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)- piper-azin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661.
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel ki-nase and nonkinase targets. Blood 2007, 110, 4055–4063.
- Kantarjian, H.; Jabbour, E.; Grimley, J.; Kirkpatrick, P. Dasatinib. Nat. Rev. Drug Discov. 2006, 5, 717–718.
- Kantarjian, H.; Shah, N.P.; Hochhaus, A.; Cortes, J.; Shah, S.; Ayala, M.; Moiraghi, B.; Shen, Z.; Mayer, J.; Pasquini, R.; et al. Dasatinib versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2010, 362, 2260–2270.
- Lindauer, M.; Hochhaus, A. Small Molecules in Hematology; Springer Nature 2018; Volume 212, pp. 31–68.
- Naqvi, K.; Jabbour, E.; Skinner, J.; Anderson, K.; Dellasala, S.; Yilmaz, M.; Ferrajoli, A.; Bose, P.; Thompson, P.; Alvarado, Y.; et al. Long-term follow-up of lower dose dasatinib (50 mg daily) as frontline therapy in newly diagnosed chronic-phase chronic myeloid leukemia. Cancer 2020, 126, 67–75.
- Remsing Rix, L.L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K.L.; Stranzl, T.; Müller, A.; Baumgartner, C.; Valent, P.; Au-gustin, M.; et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 2009, 23, 477–485.
- Boschelli, D.H.; Ye, F.; Wang, Y.D.; Dutia, M.; Johnson, S.L.; Wu, B.; Miller, K.; Powell, D.W.; Yaczko, D.; Young, M.; et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J. Med. Chem. 2001, 44, 3965–3977.
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of Bosutinib, Dasatinib, and Nilotinib Against 18 Imatinib- Resistant BCR/ABL Mutants. J. Clin. Oncol. 2009, 27, 468–469.
- Gambacorti-Passerini, C.; Brummendorf, T.H.; Kim, D.-W.; Turkina, A.G.; Masszi, T.; Assouline, S.; Durrant, S.; Kantarjian, H.M.; Khoury, H.J.; Zaritskey, A.; et al. Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: Minimum 24-month follow-up. Am. J. Hematol. 2014, 89, 732–742.
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Mi-lojkovic, D.; Le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237.
- Rusconi, F.; Piazza, R.; Vagge, E.; Gambacorti-Passerini, C. Bosutinib: A review of preclinical and clinical studies in chronic myelogenous leukemia. Expert Opin. Pharmacother. 2014, 15, 701–710.
- Hochhaus, A.; Gambacorti-Passerini, C.; Abboud, C.; Gjertsen, B.T.; Brümmendorf, T.H.; Smith, B.D.; Ernst, T.; Giral-do-Castellano, P.; Olsson-Strömberg, U.; Saussele, S.; et al. Bosutinib for pretreated patients with chronic phase chronic mye-loid leukemia: Primary results of the phase 4 BYOND study. Leukemia 2020, 34, 2125–2137.
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.; Leoni, V.; e t al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol. 2018, 5, e641–e652.
- Li, S. Src-family kinases in the development and therapy of Philadelphia chromosome-positive chronic myeloid leukemia and acute lymphoblastic leukemia. Leuk. Lymphoma 2008, 49, 19–26.
- Hu, Y.; Swerdlow, S.; Duffy, T.M.; Weinmann, R.; Lee, F.Y.; Li, S. Targeting multiple kinase pathways in leukemic progeni-tors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 16870–16875.
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541.
- De Keersmaecker, K.; Porcu, M.; Cox, L.; Girardi, T.; Vandepoel, R.; De Beeck, J.O.; Gielen, O.; Mentens, N.; Bennett, K.L.; Oliver Hantsche, O. NUP214-ABL1-mediated cell proliferation in T-cell acute lymphoblastic leukemia is dependent on the LCK kinase and various interacting proteins. Haematologica 2014, 99, 85–93.
- Deenik, W.; Beverloo, H.B.; van der Poel-van de Luytgaarde, S.C.P.A.M.; Wattel, M.M.; van Esser, J.W.J.; Valk, P.J.M.; Cor-nelissen, J.J. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 627–629.
- Divakar, S.K.A.; Ramana Reddy, M.V.; Cosenza, S.C.; Baker, S.J.; Perumal, D.; Antonelli, A.C.; Brody, J.; Akula, B.; Parekh, S., Premkumar Reddy, E. Dual inhibition of CDK4/Rb and PI3K/AKT/mTOR pathways by ON123300 induces synthetic le-thality in mantle cell lymphomas. Leukemia 2016, 30, 86–89.
- Natoni, A.; Murillo, L.S.; Kliszczak, A.E.; Catherwood, M.A.; Montagnoli, A.; Samali, A.; O’Dwyer, M.; Santocanale, C. Mechanisms of action of a dual Cdc7/Cdk9 kinase inhibitor against quiescent and proliferating CLL cells. Mol. Cancer Ther. 2011, 10, 1624–1634.
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365.
- Natoni, A.; Coyne, M.R.E.; Jacobsen, A.; Rainey, M.D.; O’Brien, G.; Healy, S.; Montagnoli, A.; Moll, J.; O’Dwyer, M.; Santo-canale, C. Characterization of a dual CDC7/CDK9 inhibitor in multiple myeloma cellular models. Cancers 2013, 5, 901–918.
- O’ Reilly, E.; Dhami, S.P.S.; Baev, D. V.; Ortutay, C.; Halpin-McCormick, A.; Morrell, R.; Santocanale, C.; Samali, A.; Quinn, J.; O'Dwyer, M.E.; et al. Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML. Sci. Rep. 2018, 8, 15752.
- McLaughlin, R.P.; He, J.; Van Der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.M.; Smid, M.; Zhang, Y.; van de Water, B. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019, 21, 77.
- Chen, E.W.; Tay, N.Q.; Brzostek, J.; Gascoigne, N.R.J.; Rybakin, V. A Dual Inhibitor of Cdc7/Cdk9 Potently Suppresses T Cell Activation. Front. Immunol. 2019, 10, 1718.
- Steele, A.J.; Prentice, A.G.; Cwynarski, K.; Hoffbrand, A.V.; Hart, S.M.; Lowdell, M.W.; Samuel, E.R.; Wickremasinghe, R.G. The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: Potential for reversal of cytoprotection by the microenvironment. Blood 2010, 116, 4569–4577.
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402.
- Ishikawa, C.; Senba, M.; Mori, N. Anti-adult T-cell leukemia/lymphoma activity of cerdulatinib, a dual SYK/JAK kinase in-hibitor. Int. J. Oncol. 2018, 53, 1681–1690.
- Coffey, G.; Betz, A.; DeGuzman, F.; Pak, Y.; Inagaki, M.; Baker, D.C.; Hollenbach, S.J.; Pandey, A.; Sinha, U. The novel ki-nase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-Cell cancer. J. Pharmacol. Exp. Ther. 2014, 351, 538–548.
- Blunt, M.D.; Koehrer, S.; Dobson, R.C.; Larrayoz, M.; Wilmore, S.; Hayman, A.; Parnell, J.; Smith, L.D.; Davies, A., Johnson, P.W.M.; et al. The dual Syk/JAK inhibitor cerdulatinib antagonizes b-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin. Cancer Res. 2017, 23, 2313–2324.
- Coffey, G.P.; Feng, J.; Betz, A.; Pandey, A.; Birrell, M.; Leeds, J.M.; Der, K.; Kadri, S.; Lu, P.; Segal, J.P.; et al. Cerdulatinib pharmacodynamics and relationships to tumor response following oral dosing in patients with relapsed/refractory b-cell ma-lignancies. Clin. Cancer Res. 2019, 25, 1174–1184.
- Hamlin, P.A.; Flinn, I.W.; Wagner-Johnston, N.; Burger, J.A.; Coffey, G.P.; Conley, P.B.; Michelson, G.; Leeds, J.M.; Der Bs, K.; Kim, Y.; et al. Efficacy and safety of the dual SYK/JAK inhibitor cerdulatinib in patients with relapsed or refractory B-cell malignancies: Results of a phase I study. Am. J. Hematol. 2019, 94, E90–E93.
- Wullenkord, R.; Friedrichs, B.; Erdmann, T.; Lenz, G. Therapeutic potential of PI3K signaling in distinct entities of B-cell lymphoma. Expert Rev. Hematol. 2019, 12, 1053–1062.
- Okkenhaug, K. Two birds with one stone: Dual p110δ and p110γ inhibition. Chem. Biol. 2013, 20, 1309–1310.
- Winkler, D.G.; Faia, K.L.; Dinitto, J.P.; Ali, J.A.; White, K.F.; Brophy, E.E.; Pink, M.M.; Proctor, J.L.; Lussier, J.; Martin, C.M.; et al. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and in-flammatory disease models. Chem. Biol. 2013, 20, 1364–1374.
- Flinn, I.W.; Miller, C.B.; Ardeshna, K.M.; Tetreault, S.; Assouline, S.E.; Mayer, J.; Merli, M.; Lunin, S.D.; Pettitt, A.R.; Nagy, Z.; et al. DYNAMO: A Phase II study of duvelisib (IPI-145) in patients with refractory indolent non-hodgkin lymphoma. J. Clin. Oncol. 2019, 37, 912–922.
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455.
- Liu, X.; Wang, A.; Liang, X.; Liu, J.; Zou, F.; Chen, C.; Zhao, Z.; Deng, Y.; Wu, H.; Qi, Z.; et al. Simultaneous inhibition of Vps34 kinase would enhance PI3Kδ inhibitor cytotoxicity in the B-cell malignancies. Oncotarget 2016, 7, 53515–53525.
- Maharaj, K.; Powers, J.J.; Achille, A.; Mediavilla-Varela, M.; Gamal, W.; Burger, K.L.; Fonseca, R.; Jiang, K.; Miskin, H.P.; Maryanski, D.; et al. The dual PI3Kd/CK1e inhibitor umbralisib exhibits unique immunomodulatory effects on CLL T cells. Blood Adv. 2020, 4, 3072–3084.
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.-X.; et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kδ and CK1ε in hematological malignancies. Blood 2017, 129, 88–99.
- Burris, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: An open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018, 19, 486–496.
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 is a novel dual PI3K/mTOR inhibitor with preclinical antitumor activity in lymphomas as a single agent and in combination therapy. Clin. Cancer Res. 2018, 24, 120–129.
- Tarantelli, C.; Lupia, A.; Stathis, A.; Bertoni, F. Is there a role for dual PI3K/mTOR inhibitors for patients affected with lym-phoma? Int. J. Mol. Sci. 2020, 21, 1060.
- Beaufils, F.; Cmiljanovic, N.; Cmiljanovic, V.; Bohnacker, T.; Melone, A.; Marone, R.; Jackson, E.; Zhang, X.; Sele, A.; Borsari, C.; et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class i PI3K/mTOR Inhibitor as Clinical Candidate in Oncology. J. Med. Chem. 2017, 60, 7524–7538.
- Collins, G.P.; Popat, R.; Stathis, A.; Krasniqi, F.; Eyre, T.A.; Ng, C.H.; El-Sharkawi, D.; Schmidt, C.; Wicki, A.; Ivanova, E.; et al. A Dose-Escalation (DE) Study with Expansion Evaluating Safety, Pharmacokinetics and Efficacy of the Novel, Balanced PI3K/mTOR Inhibitor PQR309 in Patients with Relapsed or Refractory Lymphoma. Blood 2016, 128, 5893.
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16.
- Tasian, S.K.; Teachey, D.T.; Li, Y.; Shen, F.; Harvey, R.C.; Chen, I.M.; Ryan, T.; Vincent, T.L.; Willman, C.L.; Perl, A.E.; et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lympho-blastic leukemia. Blood 2017, 129, 177–187.
- Wong, J.; Welschinger, R.; Hewson, J.; Bradstock, K.F.; Bendall, L.J. Efficacy of dual PI-3K and mTOR inhibitors in Vitro and in Vivo in acute lymphoblastic leukemia. Oncotarget 2014, 5, 10460–10472.
- Chiarini, F.; Grimaldi, C.; Ricci, F.; Tazzari, P.L.; Evangelisti, C.; Ognibene, A.; Battistelli, M.; Falcieri, E.; Melchionda, F.; Pession, A.; et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010, 70, 8097–8107.
- Schult, C.; Dahlhaus, M.; Glass, A.; Fischer, K.; Lange, S.; Freund, M.; Junghanss, C. The dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exerts anti-proliferative activity towards acute lymphoblastic leukemia cells. Anticancer Res. 2012, 32, 463–474.
- Bhende, P.M.; Park, S.I.; Lim, M.S.; Dittmer, D.P.; Damania, B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia 2010, 24, 1781–1784.
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26.
- Chiarini, F.; Falà, F.; Tazzari, P.L.; Rlcci, F.; Astolfi, A.; Pession, A.; Pagliaro, P.; McCubrey, J.A.; Martelli, A.M. Dual inhibi-tion of class ia phosphatidyl inositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009, 69, 3520–3528.
- Thijssen, R.; Ter Burg, J.; Garrick, B.; Van Bochove, G.G.W.; Brown, J.R.; Fernandes, S.M.; Rodríguez, M.S.; Michot, J.-M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583.
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-in-human phase I study of a dual mTOR kinase and DNA-PK inhibitor (CC-115) in advanced malignancy. Cancer Manag. Res. 2019, 11, 10463–10476.
- Bressanin, D.; Evangelisti, C.; Ricci, F.; Tabellini, G.; Chiarini, F.; Tazzari, P.L.; Melchionda, F.; Buontempo, F.; Pagliaro, P.; Pession, A.; et al. Harnessing the PI3K/Akt/mtor pathway in T-cell acute lymphoblastic leukemia: Eliminating activity by tar-geting at different levels. Oncotarget 2012, 3, 811–823.
- Jerkeman, M.; Hallek, M.; Dreyling, M.; Thieblemont, C.; Kimby, E.; Staudt, L. Targeting of B-cell receptor signalling in B-cell malignancies. J. Intern. Med. 2017, 282, 415–428.
- Liu, L.; Shi, B.; Li, X.; Wang, X.; Lu, X.; Cai, X.; Huang, A.; Luo, G.; You, Q.; Xiang, H. Design and synthesis of benzofu-ro[3,2-b]pyridin-2(1H)-one derivatives as anti-leukemia agents by inhibiting Btk and PI3Kδ. Bioorg. Med. Chem. 2018, 26, 4537–4543.
- Liu, L.; Li, X.; Cheng, Y.; Wang, L.; Yang, H.; Li, J.; He, S.; Shuangjie, W.; Yin, Q.; Xiang, H. Optimization of novel benzofu-ro[3,2-b]pyridin-2(1H)-one derivatives as dual inhibitors of BTK and PI3Kδ. Eur. J. Med. Chem. 2019, 164, 304–316.
- Wu, H.; Hu, C.; Wang, A.; Weisberg, E.L.; Chen, Y.; Yun, C.H.; Wang, W.; Liu, Y.; Liu, X.; Tian, B.; et al. Discovery of a BTK/MNK dual inhibitor for lymphoma and leukemia. Leukemia 2016, 30, 173–181.
- Abaza, Y.; Kantarjian, H.; Alwash, Y.; Borthakur, G.; Champlin, R.; Kadia, T.; Garcia-Manero, G.; Daver, N.; Ravandi, F.; Verstovsek, S; et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am. J. Hematol. 2020, 95, 1288-1295.
- Mologni, L.; Cleris, L.; Magistroni, V.; Piazza, R.; Boschelli, F.; Formelli, F.; Gambacorti‐Passerini, C. Valproic acid enhances bosutinib cytotoxicity in colon cancer cells. Int. J. Cancer 2009, 124, 1990–1996.
- Vancurova, I.; Uddin, M.M.; Zou, Y.; Vancura, A. Combination Therapies Targeting HDAC and IKK in Solid Tumors. Trends Pharmacol. Sci. 2018, 39, 295–306.
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323.
- Mondello, P.; Derenzini, E.; Asgari, Z.; Philip, J.; Brea, E.J.; Seshan, V.; Hendrickson, R.C.; De Stanchina, E.; Scheinberg, D.A.; Younes, A. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget 2017, 8, 14017–14028.
- Guo, H.; Zeng, D.; Zhang, H.; Bell, T.; Yao, J.; Liu, Y.; Huang, S.; Li, C.J.; Lorence, E.; Zhou, S.; et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 2019, 38, 1802–1814.
- Ishikawa, C.; Mori, N. The role of CUDC-907, a dual phosphoinositide-3 kinase and histone deacetylase inhibitor, in inhibit-ing proliferation of adult T-cell leukemia. Eur. J. Haematol. 2020, 105, 763–772.
- Younes, A.; Berdeja, J.G.; Patel, M.R.; Flinn, I.; Gerecitano, J.F.; Neelapu, S.S.; Kelly, K.R.; Copeland, A.R.; Akins, A.; Clancy, M.S.; et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17, 622–631.
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.S.; Ma, A.W.; Xu, G.-X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer Ther. 2017, 16, 285–299.
- Chen, Y.; Peubez, C.; Smith, V.; Xiong, S.; Kocsis-Fodor, G.; Kennedy, B.; Wagner, S.; Balotis, C.; Jayne, S., Dyer, M.J.S.; et al. CUDC-907 blocks multiple pro-survival signals and abrogates microenvironment protection in CLL. J. Cell Mol. Med. 2019, 23, 340–348.
- Li, X.; Su, Y.; Madlambayan, G.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Ma, J.; Knight, T.; et al. Antileu-kemic activity and mechanism of action of the novel PI3K and histone deacetylase dual inhibitor CUDC-907 in acute myeloid leukemia. Haematologica 2019, 104, 2225–2240.
- Huang, Y.; Dong, G.; Li, H.; Liu, N.; Zhang, W.; Sheng, C. Discovery of Janus Kinase 2 (JAK2) and Histone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for the Combinational Treatment of Leukemia and Invasive Fungal Infections. J. Med. Chem. 2018, 61, 6056–6074.
- Kim, S.; Min, A.; Lee, K.H.; Yang, Y.; Kim, T.Y.; Lim, J.M.; Park, S.J.; Nam, H.-J.; Kim, J.E.; Song, S.-H. Antitumor effect of KX-01 through inhibiting Src family kinases and mitosis. Cancer Res. Treat. 2017, 49, 643–655.
- Malik, H.S.; Bilal, A.; Ullah, R.; Iqbal, M.; Khan, S.; Ahmed, I.; Krohn, K.; Saleem, R.S.; Hussain, H.; Faisal, A. Natural and Semisynthetic Chalcones as Dual FLT3 and Microtubule Polymerization Inhibitors. J. Nat. Prod. 2020, 83, 3111–3121.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Burslem, G.M.; Crews, C.M. Small-Molecule Modulation of Protein Homeostasis. Chem. Rev. 2017, 117, 11269–11301.
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190.
- Raina, K.; Crews, C.M. Targeted protein knockdown using small molecule degraders. Curr. Opin. Chem. Biol. 2017, 39, 46–53.
- Fisher, S.L.; Phillips, A.J. Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol. 2018, 44, 47–55.
- Wang, P.; Zhou, J. Proteolysis Targeting Chimera (PROTAC): A Paradigm-Shifting Approach in Small Molecule Drug Dis-covery. Curr. Top. Med. Chem. 2018, 18, 1354–1356.
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50.
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30.
- Paiva, S.-L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119.
- Menzl, I.; Zhang, T.; Berger-Becvar, A.; Grausenburger, R.; Heller, G.; Prchal-Murphy, M.; Edlinger, L.; Knab, V.M.; Uras, I.Z.; Grundschober, E.; et al. A kinase-independent role for CDK8 in BCR-ABL1+ leukemia. Nat. Commun. 2019, 10, 4741.
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032.
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.-Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573.
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 In-hibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432.
- Mu, X.; Bai, L.; Xu, Y.; Wang, J.; Lu, H. Protein targeting chimeric molecules specific for dual bromodomain 4 (BRD4) and Polo-like kinase 1 (PLK1) proteins in acute myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2020, 521, 833–839.
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255.
- Zhang, C.; Han, X.-R.; Yang, X.; Jiang, B.; Liu, J.; Xiong, Y.; Jin, J. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304–314.
- Kang, C.H.; Lee, D.H.; Lee, C.O.; Du Ha, J.; Park, C.H.; Hwang, J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 2018, 505, 542–547



