 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Celso Mello | + 3915 word(s) | 3915 | 2021-02-03 08:45:19 | | | |
| 2 | Vivi Li | + 4 word(s) | 3919 | 2021-02-05 04:51:30 | | |
Video Upload Options
Desmoplastic small round cell tumor (DSRCT) is an extremely rare, aggressive sarcoma affecting adolescents and young adults with male predominance. Generally, it originates from the serosal surface of the abdominal cavity. The hallmark characteristic of DSRCT is the EWSR1–WT1 gene fusion. This translocation up-regulates the expression of PDGFRα, VEGF and other proteins related to tumor and vascular cell proliferation. Current management of DSRCT includes a combination of chemotherapy, radiation and aggressive cytoreductive surgery plus intra-peritoneal hyperthermic chemotherapy (HIPEC). Despite advances in multimodal therapy, outcomes remain poor since the majority of patients present disease recurrence and die within three years. The dismal survival makes DSRCT an orphan disease with an urgent need for new drugs. The treatment of advanced and recurrent disease with tyrosine kinase inhibitors, such as pazopanib, sunitinib, and mTOR inhibitors was evaluated by small trials.
1. Introduction
Desmoplastic small round cell tumor (DSRCT) is an extremely rare, aggressive sarcoma. It affects mainly adolescents and young adults and originates in and primarily involves the serosal surfaces of the abdominal cavity. It was first described by Gerald and Rosai in 1989 as a newly characterized clinicopathologic entity [1]. Current management of the disease includes a combination of chemotherapy, radiation, and aggressive surgical resection [2][3] as summarized in Figure 1. Despite advances in multimodal therapy, the outcome remains poor since the majority of patients develop high rates of disease recurrence or die within three years [4][5]. Due to the dismal survival, DSRCT has an urgent unmet need for more effective and innovative therapeutic options.
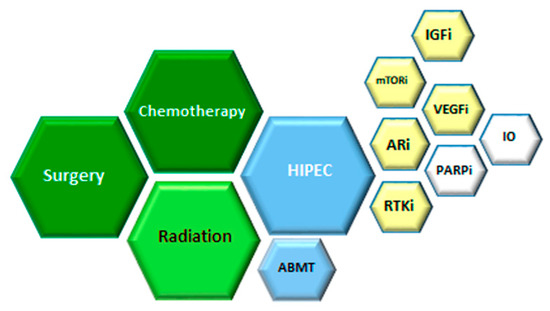
Figure 1. Therapeutic options for desmoplastic small round cell tumor (DSRCT). Based on multiple retrospectives and few prospective studies, the benefit was observed for therapies in green, low evidence of the benefit for therapies in blue. For relapsed or progressive disease, strategies in yellow had been used and in white are perspectives. HIPEC (hyperthermic intraperitoneal chemotherapy), RTKi (Receptor Tyrosine Kinase inhibition), ARi (Androgen Receptor inhibition), VEGFi (Vascular Endothelial Growth Factor inhibition), IO (immune check point inhibition), IGFi (Insulin Growth Factor inhibiotion), mTORi (mammalian Target of Rapamycin inhibition), ABMT (autologous bone marrow transplant).
2. Clinical Presentation and Diagnosis
The disease predominantly originates from the peritoneum or retroperitoneum and can invade the omentum with multiple peritoneal implants involving the diaphragm, splenic hilum, mesentery of the small and large bowel, and the pelvic peritoneum [2][6][7]. Patients can be asymptomatic for long periods of time until symptoms of pain, ascites, constipation, weight loss, distension and jaundice [2][6]. Other sites of the primary tumor are described in the literature as the thoracic cavity, testicle, head and neck, intracranial, thigh, axilla/shoulder, intraosseous, uterine corpus, ovary, skull, middle ear, and others [8][9][10][11][12]. About one-half of the patients will present extra-peritoneal metastasis at the time of the diagnosis [13][6][14][15], although this percentage was lower in the report of Stiles et al. [7]. The liver and lung are the two most common sites for distant metastasis [2][3][7][14].
In most cases, patients with an abdominal disease are diagnosed in an advanced stage, with large masses and/or extensive seeding in the visceral and parietal peritoneum [15][16]. Symptoms, which are related to the tumor burden and location of the lesions, motivate investigation by image exams. The most common imaging finding is multiple, lobulated, low-attenuated, heterogeneous peritoneal, omental and serosal soft tissue masses usually discrete, round or ovoid, without an apparent primary organ of origin [17][18][19]. Almost all patients will present a dominant mass, mainly in the retrovesical or recto-uterine location, peritoneal or omental [18]. MRI can be helpful in delineating the extent of the disease, if surgical resection is considered [20] and can reveal lesions with heterogeneous contrast enhancement [21]. The role for position emission tomography (PET)-CT is not well established in DSRCT imaging, although it has been used each more as part of staging evaluation together with a chest CT scan [16][21]. There is no formal staging system for DSRCT [22]. One was proposed by Hayes-Jordan et al. [23] using the Peritoneal Cancer Index but it has not yet been validated.
Histologically, the tumor consists of solid sheets, large nests, small clumps, or cords of cohesive, small, round, ovoid, or spindled cells, with inconspicuous nucleoli, and scant cytoplasm, lying in a hypocellular, desmoplastic, collagenous stroma [24]. In immunohistochemistry, there is an expression of desmin, membrane antigen (EMA), cytokeratins (AE1/AE3 and CAM5.2) and neural markers (neuron-specific enolase and CD57), and smaller numbers expressing chromogranin, synaptophysin, CD56, neurofilament protein and S100 protein [25]. DSRCT can be immunoreactive for antibodies selectively directed to the carboxy terminus of the WT1 protein in more than 90% of cases [26]. It is important to note that DSRCT can show a polyphenotypic immune profile as well as a marked variation in morphologic appearances from tumor to tumor and within the same neoplasm [27].
3. Treatment
3.1. Therapeutic Approach for Newly Diagnosed Patients
3.1.1. Radiation Therapy
Given the propensity for morbid intra-peritoneal progression, consolidative whole abdominopelvic radiotherapy (WAP-RT) as part of multimodal treatment (chemotherapy, surgical debulking and WAP-RT) was first reported using the P6 protocol, in an attempt to improve local control [28]. Honoré et al. [14], reported in a series of 38 patients with a median follow-up of almost 5 years, that multimodal treatment combining systemic chemotherapy, complete macroscopic resection, and postoperative WAP-RT could prolong survival in patients without the extra-peritoneal disease (EPM)—median survival of 37.7 months (range 7.9–42.9 months). The factors predictive of 3-year overall survival were the absence of EPM, complete surgical resection, postoperative WAP-RT and postoperative chemotherapy [14].
Atallah et al. [29] studied the prognostic role of WAP-RT on oncologic outcomes as part of multimodal treatment in 103 patients with abdominal DSRCT treated at eight French centers from 1991 to 2014. Patients were retrospectively divided into three groups for evaluation: Group A treated with adjuvant RT after cytoreductive surgery, Group B without RT after cytoreductive surgery, and Group C treated with chemotherapy alone. Three-year OS was 61.2% (range 41–76%) in Group A, 37.6% (range 22–53.1%) in Group B, and 17.3% (range 6.3–32.8%) for Group C, respectively (p < 0.001). Peritoneal progression-free survival (PPFS) and progression-free survival (PFS) also differed significantly between the three groups (p < 0.001), but not distant progression. They concluded that RT seems to improve survival after cytoreductive surgery, with better PPFS, PFS and OS for the patients treated in a multimodal approach, with the limitations of a retrospective study, lack of statistical power (due to the small number of patients), and the need of randomized prospective studies to confirm these results [29].
In a more recent publication, Subbiah et al. [30] reported the MDAnderson Cancer Center experience with the treatment of 187 patients over two decades with the multimodal approach. The five-year OS rate was substantially improved from 5% to 25% with newer chemotherapy agents and better surgical and RT techniques. Chemotherapy response and complete cytoreductive surgery (CCS) were associated with improved survival. Their results also supported the use of WAP-RT when the time of diagnosis was used as a reference to estimate OS (univariate analysis, p = 0.01; HR, 0.44) [30]. However, because RT was given almost exclusively to patients who underwent CCS after chemotherapy, they removed these confounding factors and assessed the effects of WAP-RT using the date of the surgery as the start date in a time-variant analysis, and surprisingly, WAP-RT did not improve OS. This unexpected result conflicts with current clinical practice of a tri-modality therapy (chemo, surgery and radiation), and updated their treatment recommendation to consider WAP-RT in highly selected patients that are prospectively monitored in clinical trials [30]. Desai et al. [31] reported that acute toxicities of WAP-RT were primarily gastrointestinal and hematologic, and were improved in comparisons of IMRT against 2D-RT (gastrointestinal grade 2 or higher: 33% × 77%, p = 0.04; and grade 4 hematologic: 33% × 82%, p = 0.02), with no survival differences. Late toxicity (small bowel obstruction) did not statistically differ between RT modalities [31]. There are two main indications of radiation therapy in the control of DSRCT. One is in the palliative setting and the other is in an adjuvant scenario after complete cytoreductive surgery.
3.1.2. HIPEC
Due to the rarity of this disease and analysis based on retrospective series, the role of radiotherapy in the management of DSRCT is still controversial. Appropriate patient selection is critical, as severe toxicities can occur. Despite aggressive multidisciplinary approaches, patients have a poor prognosis. Prospective randomized multicenter studies will be needed to evaluate the role of local treatments such as RT in the course of the disease.
Even after chemotherapeutic cytoreduction and surgical resection of gross, visible disease, the microscopic residual disease is often present [4]. Hence, hyperthermic intra-peritoneal chemotherapy (HIPEC) has been examined as an adjunctive intraoperative strategy. In a recent phase 2 trial, 14 DSRCT patients were treated with neoadjuvant chemotherapy, followed by cytoreductive surgery (CRS), which was complete (CR0) or near- complete (CR1 ≤ 2.5 cm of tumor remaining) in all patients, with closed technique HIPEC using 100 mg/m2 of cisplatin for 90 min at 41 degrees Celsius, then followed by WAP-RT [32]. The 3-year overall survival from time of diagnosis for DSRCT patients was 79%, and the estimated median recurrence-free survival (RFS) was 14.0 months. In 100% of patients without hepatic or portal metastasis, there was no peritoneal disease recurrence after CRS-HIPEC. They concluded that CRS, HIPEC and WART are effective local control therapy in DSRCT patients. Earlier, it was demonstrated that patients who had CR0 or CR1 and HIPEC had significantly longer median survival compared with patients who had HIPEC and gross residual disease greater than 2.5 cm after surgical cytoreduction (63.4 vs. 26.7 months) [33]. In an important retrospective study conducted at MDAnderson Cancer Center, most of the patients (72%) received combined treatment with chemotherapy, surgery plus HIPEC and radiation therapy. This study was underpowered to conclude about the impact of HIPEC on the OS. However, there was not found improvement in the three- and five-year OS (p = 0.12) of patients treated with HIPEC. Patients with DSRCT and disease outside the abdomen at the time of surgery do not benefit from HIPEC [33]. Until now, there is no randomized trials designed to evaluate the relative contribution to improve outcome after complete surgical excision of intra-abdominal implants.
A retrospective study with 187 DSRCT patients confirmed that chemotherapy and CCS remain the cornerstone of treatment, and suggest that prospective randomized studies will be required to prove the unequivocal benefit of HIPEC or WAP RT in the management of DSRCT [30].
3.3. Current and Emerging Therapy for Relapsed or Progressive Disease
DSRCT is characterized by poor response to conventional chemotherapy and early relapse after radical surgery. Second-line treatment is ineffective in most cases. In our cohort, out of 19 patients treated with first-line chemotherapy, 13 received the second-line and the progression-free survival was only 3.9 months [34]. This short survival time highlights the aggressiveness of this disease and the challenge in developing new therapeutic strategies to treat these young patients. Despite the development of new regimens for ES and other soft tissue and bone sarcoma in recent years, DSRCT is underrepresented or were not included in the trials that lead to the drug approval [35]. As a result, the evidence to use second-line therapy is very limited. It is paramount to develop active cooperative groups to quickly collect data and propose new strategies for the treatment of DSRCT. Moreover, patients outside Europe and North America are almost never offered the opportunity to participate in clinical trials for rare diseases. The SELNET (https://selnet-h2020.org/), that is a Horizon 2020/EU project, aims to create a network between European and Latin American countries to improve diagnosis and treatment of sarcomas and eventually to develop clinical trials in these centers across the Atlantic Ocean.
3.3.1. The Importance of Pre-Clinical Models to Drug Development for Rare Sarcoma
The use of preclinical models is an important step in the development of new therapies for tumors in general and is crucial for rare tumors such as DSRCT. Due to the rarity of DSRCT, conducting clinical trials with new drugs is extremely difficult for many reasons [36]. First, the fact that genetic and functional comprehensive analyses of these tumors are limited. Second, the aggressive behavior and chemotherapy resistance makes the inclusion in target therapy clinical trials difficult. Accrual of patients in clinical trials is difficult due to the limited number of individuals affected yearly. As a result, the use of pre-clinical models is an important step in the development of novel drugs since a higher number of mechanisms can be modulated and faster therapeutic targets can be explored. Modeling these tumors with experimental models allows the investigation of the molecular mechanisms that underlie tumor origin and progression. The fidelity of the model is also related to the predictive capacity to anticipate eventual effects of drugs which will help to determine the efficiency and efficacy of anti-tumoral drugs. In the case of rare tumors, the possibility of scaling-up is crucial to translate technology from the bench to the bedside.
In 2002, Nishio and collaborators [37] reported for the first time the development of the DSRCT cell line, named JN-DSRCT-1, derived from pleural effusion from a 7-year-old patient with lung metastasis. JN-DSRCT-1 cells are small, round or spindle-shaped with oval nuclei and were maintained continuously in vitro for more than 190 passages for more than 40 months. The cell has a tumorigenic capacity and the histology of heterotransplanted tumors in SCID mice maintains the characteristics of the original DSRCT, including the expression of immunohistochemical markers (vimentin, desmin, CD57, among others), a t(11; 22) translocation (p13; q12) and presence of EWS-WT1 fusion [37]. JN-DSRCT-1 cells are being used in several studies that help to unveil DSRCT biology, especially the role of EWS-WT1 fusion protein and also to identify targets for therapeutic interventions [38][39].
As mentioned before, EWS-WT1 translocation is the major driver in DSRCT and plays many roles in tumor biology that have the potential to be used as therapeutic targets. As already described in other studies, EWS-WT1 gene underwent RNA splicing and one variant lacks three amino-acids and was named EWS-WT1(-KTS) due to the absence of Lys-Thr-Ser residues [40]. This isoform activates a gene encoding a tetraspanin-family protein, T-cell acute lymphoblastic leukemia-associated antigen 1 (TALLA-1) [41]. TALLA-1 is part of multi-protein family involved in several processes, such as cell adhesion, migration and metastasis, and this gene could be a candidate for diagnostic marker and a putative target for therapy [41]. Another target for EWS-WT1 fusion gene is ENT4 (equilibrative nucleoside transporter 4) which encodes a pH-dependent adenosine transporter [42]. Neural gene induction is also triggered by EWS-WT1 in JN-DSRCT-1 cells and neural reprogramming factor ASCL1 is an important player in mediating multiple WT1-responsive elements, suggesting that neural differentiation pathway could be tested as therapeutic agents for DSRCT [43]. Recent work described the dependence of EWS-WT1 in DSRCT survival [38]. Silencing EWS-WT1 causes proliferation loss, growth arrest and gene expression analysis indicates repression of estrogen signaling and highlights therapeutic genetic vulnerabilities, such as FGFR4, JAK3, mTOR, PDGF, ERG, and TGFB1 genes [38]. Another study that evaluated potential therapeutic targets performed RNA sequencing of 12 tumor samples from pediatric patients with DSRCT found high expression of IGF2, FGFR4, CD200 and CD276, the latter two molecules are candidates for immune checkpoint inhibitor therapy [44].
In terms of therapy, the first use of JN-DSRCT-1 cells was to test the effect of rapamycin-induced apoptotic death [45]. The mechanism involves the up-regulation of Bax concomitant Bcl-xL down-regulation. Rapamycin also down-regulates EWS-WT1 and 26S p44.5 proteasome subunit, suggesting that rapamycin induces apoptosis by preventing the degradation of the Bax protein by the proteasome, and that this process is independent of mTOR inhibition. Furthermore, these results strongly support the introduction of the use of rapamycin as a cytotoxic agent for the treatment of DSRCT. JN-DSRCT-1 cells were tested to verify the effectiveness of the TRAIL receptor agonist (apoptosis inducer), called ONC201 [46]. In this study, it was found that the induction of TRAIL decreases proliferation and induces apoptosis in vitro and decreases tumor growth in vivo. The potential of anti-angiogenic agents in decreasing tumor growth of the JN-DSRCT-1 cell was also investigated [36]. Animals with JN-DSRCT-1 cell xenografts were treated with bevacizumab and showed a prolongation of the time to progression and the long-term regressions were marked after treatment with the combination of irinotecan and bevacizumab compared to irinotecan alone [36]. Interestingly, there is recent evidence that indicates that the use of other anti-angiogenic agents may be effective in the treatment of DSRCT, as in the case report of a patient with advanced DSRCT, in the second-line of treatment, refractory to cisplatin, was treated with apatinib (VEGFR-2 inhibitor drug) and had a positive response, with a significant reduction in tumor mass [47]. JN-DSRCT-1 cells are sensitive to alkylating agent trabectedin and the mechanism of action involves the expression of genes involved with proliferation and apoptosis [48]. An alternative mechanism of action of trabectedin is the impairment of transactivation of FUS-CHOP fusion protein in liposarcoma [49]. This activity is also observed in EWS-WT1 fusion protein, trabectedin reduces its binding on its target gene promoters and, thus, affects EWS-WT1—dependent gene expression in JN-DSRCT-1 cells [48]. Recently, the combination of PARP inhibitor olaparib with the alkylating agent temozolomide was tested in JN-DSRCT-1 cells in vitro and in vivo and the results indicates that the combination has synergistic effects upon cell viability, inducing cell cycle arrest which progress to apoptosis induction, causing tumor reduction [39].
Additionally to JN-DSRCT-1 cells, Markides and collaborators established more DSRCT cell lines, including BER lineage that also presents EWS-WT1 fusion protein and have similar behavior of JN-DSRCT-1 [38][50]. Together, this evidence shows the importance of obtaining tumor models to accelerate preclinical research, bring possibilities for investigating new therapeutic approaches for this rare but lethal malignancy.
3.3.2. Targeting Angiogenesis and Other TKR
As a hypervascular tumor, DSRCT is characterized by an overexpression of proteins that promote and maintain the angiogenic process necessary for continued tumor growth and proliferation. EWS-WT1 is able to induce PDGFA expression [51] and activation of the IGF1R gene [52] (Figure 2). Other tyrosine kinase receptors (TKR) expression have been found to be disrupted in DSRCT and are related to proliferation and angiogensis. VEGFR-2 and VEGFA expression was found to be markedly increased in the DSRCT tumor sample and in the human DSRCT cell line, JN-DSRCT [53].
The use of tyrosine kinase inhibitors (TKI) for VEGF, VEGFR, PDGFRα and other proteins involved in tumoral vascular proliferation has been explored in the clinical scenario [47][54][55]. Pazopanib, apatinib and sunitinib inhibit angiogenesis by abrogating the VEGF-induced phosphorylation of VEGF receptors as well as other TKRs including PDGFR, FGFR, and c-KIT, affecting downstream activation of the PI3K/AKT, PKC, and other pathways that mediate cell proliferation, migration, and survival [56].
In the PALETTE study, 369 patients were randomized to receive pazopanib 800 mg/day versus placebo [35]. Median PFS was 4.6 months (95%CI: 3.7–4.8) for pazopanib compared with 1.6 months. A combined analysis of patients with the diagnosis of DSRCT treated in the EORTC phase II study 62,043 (3 patients), EORTC phase III 62,072 (3 patients) and in a UK Pazopanib expanded access program (3 patients) was performed [54]. Data from nine patients included in this analysis revealed a median age of 30 years and all patients were males with widespread metastatic DSRCT. Four patients had one previous chemotherapy line (44%), four had 2 previous chemotherapy lines (44%) and one patient 3 (12%). The response rate was partial response (PR) in 2/9 (22%) patients, stable disease (SD) in 5/9 patients (56%) and progressive disease (PD) in 2/9 (22%) with a clinical benefit rate (PR + SD > 12 weeks) of 78%. Median PFS and OS were 9.2 (95% CI: 0–23.2) and 15.4 (95% CI: 1.5–29.3) months respectively [54]. In a relatively large, retrospective study with 29 patients treated with pazopanib, clinical benefit was observed in 62% (18/29) of patients with DSRCT (CR in 1 patient, PR in 1 patient, SD in 16 patients) and the median progression-free survival was 5.4 months [57].
Sunitinib was one of the first generation of TKIs with great inhibition of VEGF receptor 2 among other TKRs. In vascular sarcomas, sunitinib showed early and promising activity in alveolar sarcoma, a chemo-resistant subtype of sarcoma [58]. In a retrospective analysis of patients with DSRCT, sunitinib showed clinical benefit in 8 patients evaluated [59]. Partial response was observed in two patients and SD in 3 patients treated in the second-and-beyond-line of therapy. Sorafenib was used in 2 patients [55], both in the fifth-line of therapy. The best response to sorafenib was stable disease and the median progression-free survival of 3.5 and 4 months. On the other hand, as first-line treatment, apatinib was used in only one patient [47]. Apatinib is another VEGFR-2 inhibitor with demonstrated activity in gastric and other tumors [60]. Clinical benefit and tumor shrinkage were reported in one patient treated with apatinib in the first-line. The patient had not received chemotherapy previously [47]. Another report showed partial response with apatinib in combination with chemotherapy as second-line treatment [61]. Ramucirumab, a VEGFR inhibitor is been tested in combination with cyclophosfomide and vinblastine in patients with relapsed and refractory DSRCT (ClinicalTrials.gov Identifier: NCT04145349).
Bevacizumab, a VEGF-A inhibitor, was combined with irinotecan and temozolamide (ITB regimen) in the first-line treatment of DSRCT. In this single-arm pilot study, 14 out of 15 patients completed the planned treatment that comprised two cycles of ITB followed by the conventional trial P6 with VAC and IE. The response rate to ITB was 27% and no major unexpected adverse event was observed [62].
3.3.3. Targeting Androgen Receptor Pathway
The increased prevalence of DSRCT in young males motivated the investigation of the testosterone synthesis pathway in tumorigenesis of this disease. In 2007, Fine et al. [63] first demonstrated AR expression in DSRCT. In a cohort of 27 heavily pretreated patients, 37% stained positive for AR. The functionality of the pathway was demonstrated by in vitro assay that showed growth of tumor cells when stimulated by di-hydro-testosterone, and inhibition of growth by flutamide [63]. Another study performing single-sample gene set enrichment analysis found that the majority of DSRCTs were enriched for the AR signature when compared to other sarcomas, such as ES and alveolar rhabdomyosarcoma [3][64].
In the previous reported Fine et al. [63] study, six patients with AR-positive DSRCT received combined androgen blockade (CAB) with bicalutamide and leuprorelin. Three patients had clinical tumor benefits for a period lasting 3 to 4 months. All of them had normal testosterone levels at the initiation of CAB therapy, while the other three non-responders had castrated levels. In another report, a patient with strong AR expression received anti-androgen therapy with bicalutamide, presenting progressive disease 2 months later [65]. Negri et al. [51], using whole genome gene expression profiling and a cancer stem cell gene array, showed that AR-positive DSRCT cells harbor characteristics of stemness, which could explain the limited effectiveness of targeting this pathway.
3.3.4. Targeting PI3K/AKT/mTOR Pathway
Activation of the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt)-mammalian target of rapamycin (mTOR) pathway is proposed to be implicated in the development of a variety of sarcomas [66][67][68]. There is emerging data indicating the possible involvement of the mTOR pathway in DSRCT. A single-center small study attempted to evaluate the morphoproteomic profiling of the mTOR pathway in DSRCT, ES and Wilms’ tumor, and showed that the PI3K/Akt/mTOR pathway is constitutively activated in DSRCT [69]. Another study described a patient with DSRCT harboring a secondary somatic mutation in the PIK3CA gene [70].
In vitro study demonstrated that rapamycin, an mTOR inhibitor, induced the apoptotic death of DSRCT line cells [45]. There are few data on the clinical efficacy of inhibition of PI3K/AKT/mTOR pathway. In one case report, a 21-year-old man with DSRCT achieved stable disease with temsirolimus for 40 weeks [65]. Tarek et al. [71] reported their experience with five patients with relapsed DSRCT treated with vinorelbine, cyclophosphamide and temsirolimus, all of them presenting partial response, with median time to progression of 8.5 months (range 7–16 months). A phase I trial evaluated the combination of cixutumumab (an IGFR antibody) with temsirolimus, which resulted in stable disease lasting longer than 5 months in two of the three patients with DSRCT of the study [72]. In a retrospective series of patients with high-grade STS treated with pazopanib plus sirolimus following progression on pazopanib, one patient with DSRCT had stable disease for 11 months with the combination treatment [73]. Recently, a trial was designed to estimate the response rate to two initial courses of temsirolimus, temozolomide and irinotecan (window therapy) in previously untreated patients with high-risk ES family of tumors, including DSRCT (ClinicalTrials.gov Identifier: NCT01946529). The interim analysis determined the window therapy did not meet the anticipated response, and trial accrual was stopped.
References
- Gerald, W.L.; Rosai, J. Case 2 Desmoplastic small cell tumor with divergent differentiation. Pediatr. Pathol. 1989, 9, 177–183.
- Dufresne, A.; Cassier, P.; Couraud, L.; Marec-Bérard, P.; Meeus, P.; Alberti, L.; Blay, J.-Y. Desmoplastic small round cell tumor: Current management and recent findings. Sarcoma 2012, 2012, 714986.
- Bulbul, A.; Fahy, B.N.; Xiu, J.; Rashad, S.; Mustafa, A.; Husain, H.; Hayes-Jordan, A. Desmoplastic Small Round Blue Cell Tumor: A Review of Treatment and Potential Therapeutic Genomic Alterations. Sarcoma 2017, 2017, 1278268.
- Hayes-Jordan, A.; Laquaglia, M.P.; Modak, S. Management of Desmoplastic Small Round Cell Tumor. Semin Pediatr. Surg. 2016, 25, 299–304.
- Honoré, C.; Delhorme, J.; Nassif, E.; Faron, M.; Ferron, G.; Bompas, E.; Glehen, O.; Italiano, A.; Bertucci, F.; Orbach, D.; et al. Can we cure patients with abdominal Desmoplastic Small Round Cell Tumor? Results of a retrospective multicentric study on 100 patients. Surg. Oncol. 2019, 29, 107–112.
- Lal, D.R.; Su, W.T.; Wolden, S.L.; Loh, K.C.; Modak, S.; La Quaglia, M.P. Results of multimodal treatment for desmoplastic small round cell tumors. J. Pediatr. Surg. 2005, 40, 251–255.
- Stiles, Z.E.; Dickson, P.V.; Glazer, E.S.; Murphy, A.J.; Davidoff, A.M.; Behrman, S.W.; Bishop, M.W.; Martin, M.G.; Deneve, J.L. Desmoplastic small round cell tumor: A nationwide study of a rare sarcoma. J. Surg. Oncol. 2018, 117, 1759–1767.
- Al-Ibraheemi, A.; Broehm, C.; Tanas, M.R.; Horvai, A.E.; Rubin, B.P.; Cheah, A.L.; Thway, K.; Fisher, C.; Bahrami, A.; Folpe, A.L.; et al. Desmoplastic Small Round Cell Tumors With Atypical Presentations: A Report of 34 Cases. Int. J. Surg. Pathol. 2018, 27, 1–8.
- Thondam, S.K.; Du Plessis, D.; Cuthbertson, D.J.; Das, K.S.V.; Javadpour, M.; Macfarlane, I.A.; Leggate, J.; Haylock, B.; Daousi, C. Intracranial desmoplastic small round cell tumor presenting as a suprasellar mass. J. Neurosurg. 2015, 122, 773–777.
- Faras, F.; Abo-Alhassan, F.; Hussain, A.; Sebire, N.; Al-Terki, A. Primary desmoplastic small round cell tumor of upper cervical lymph nodes. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2015, 120, 4–10.
- Xu, J.; Yao, M.; Yang, X.; Liu, T.; Wang, S.; Ma, D.; Li, X. Desmoplastic small round cell tumor of the middle ear. Med. Baltim. 2018, 97, 1–5.
- Nakayama, J.; Nassau, S.; Atkins, K.; Modesitt, S.C. Desmoplastic small round cell tumor of the ovary: A rare but devastating disease in young women. Gynecol. Oncol. Rep. 2014, 7, 16–18.
- Lettieri, C.K.; Garcia-Filion, P.; Hingorani, P. Incidence and Outcomes of Desmoplastic Small Round Cell Tumor: Results from the Surveillance, Epidemiology, and End Results Database. J. Cancer Epidemiol. 2014, 2014, 680126.
- Honoré, C.; Amroun, K.; Vilcot, L.; Mir, O.; Domont, J.; Terrier, P.; Le Cesne, A.; Le Pechoux, C.; Bonvalot, S. Abdominal Desmoplastic Small Round Cell Tumor: Multimodal Treatment Combining Chemotherapy, Surgery, and Radiotherapy is the Best Option. Ann. Surg. Oncol. 2015, 22, 1073–1079.
- Gani, F.; Goel, U.; Canner, J.K.; Meyer, C.F.; Johnston, F.M. A national analysis of patterns of care and outcomes for adults diagnosed with desmoplastic small round cell tumors in the United States. J. Surg. Oncol. 2019, 119, 880–886.
- Hayes-Jordan, A.; Anderson, P.M. The diagnosis and management of desmoplastic small round cell tumor: A review. Curr. Opin. Oncol. 2011, 23, 385–389.
- Bellah, R.; Suzuki-Bordalo, L.; Brecher, E.; Ginsberg, J.P.; Maris, J.; Pawel, B.R. Desmoplastic Small Round Cell Tumor in the Abdomen and Pelvis: Report of CT Findings in 11 Affected Children and Young Adults. Am. J. Roentgenol. 2013, 184, 1910–1914.
- Arora, V.C.; Price, A.P.; Fleming, S.; Sohn, M.J.; Magnan, H.; Laquaglia, M.P.; Abramson, S. Characteristic imaging features of desmoplastic small round cell tumour. Pediatr. Radiol. 2013, 43, 93–102.
- Pickhardt, P.J.; Fisher, A.J.; Balfe, D.M.; Dehner, L.P.; Huettner, P.C. Desmoplastic small round cell tumor of the abdomen: Radiologic-histopathologic correlation. Radiology 1999, 210, 633–638.
- Kis, B.; O’Regan, K.N.; Agoston, A.; Javery, O.; Jagannathan, J.; Ramaiya, N.H. Imaging of desmoplastic small round cell tumour in adults. Br. J. Radiol. 2012, 85, 187–192.
- Zhang, W.-D.; Li, C.-X.; Liu, Q.-Y.; Hu, Y.-Y.; Cao, Y.; Huang, J.-H. CT, MRI, and FDG-PET/CT imaging findings of abdominopelvic desmoplastic small round cell tumors: Correlation with histopathologic findings. Eur. J. Radiol. 2011, 80, 269–273.
- Goadsby, P.J.; Kurth, T.; Pressman, A. FDG PET/CT Imaging of Desmoplastic Small Round Cell Tumor: Findings at Staging, During Treatment and at Follow Up. Pediatr. Radiol. 2016, 35, 1252–1260.
- Hayes-Jordan, A.; Green, H.; E Fitzgerald, N.; Xiao, L.; Anderson, P.M. Novel treatment for desmoplastic small round cell tumor: Hyperthermic intraperitoneal perfusion. J. Pediatr. Surg. 2010, 45, 1000–1006.
- Lae, M.E.; Roche, P.C.; Jin, L.; Lloyd, R.V.; Nascimento, A.G. Desmoplastic Small Round Cell Tumor: A Clinicopathologic, Immunohistochemical, and Molecular Study of 32 Tumors. Am. J. Surg. Pathol. 2002, 26, 823–835.
- Ordóñez, N.G. Desmoplastic Small Round Cell Tumor: II: An Ultrastructural and Immunohistochemical Study with Emphasis on New Immunohistochemical Markers. Am. J. Surg. Pathol. 1998, 22, 1314–1327.
- Gerald, W.L.; Ladanyi, M.; De Álava, E.; Cuatrecasas, M.; Kushner, B.H.; Laquaglia, M.P.; Rosai, J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 1998, 16, 3028–3036.
- Thway, K.; Noujaim, J.; Zaidi, S.; Miah, A.B.; Benson, C.; Messiou, C.; Jones, R.L.; Fisher, C. Desmoplastic Small Round Cell Tumor: Pathology, Genetics, and Potential Therapeutic Strategies. Int. J. Surg. Pathol. 2016, 24, 672–684.
- Kushner, B.H.; Laquaglia, M.P.; Wollner, N.; A Meyers, P.; Lindsley, K.L.; Ghavimi, F.; E Merchant, T.; Boulad, F.; Cheung, N.K.; A Bonilla, M.; et al. Desmoplastic small round-cell tumor: Prolonged progression-free survival with aggressive multimodality therapy. J. Clin. Oncol. 1996, 14, 1526–1531.
- Atallah, V.; Honore, C.; Orbach, D.; Helfre, S.; Ducassou, A.; Thomas, L.; Levitchi, M.-B.; Mervoyer, A.; Naji, S.; Dupin, C.; et al. Role of adjuvant radiation therapy after surgery for abdominal desmoplastic small round cell tumors. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 1244–1253.
- Subbiah, V.; Lamhamedi-Cherradi, S.-E.; Cuglievan, B.; Menegaz, B.A.; Camacho, P.; Huh, W.W.; Ramamoorthy, V.; Anderson, P.M.; Pollock, R.E.; Lev, D.C.; et al. Multimodality treatment of desmoplastic small round cell tumor: Chemotherapy and complete cytoreductive surgery improve patient survival. Clin. Cancer Res. 2018, 24, 4865–4873.
- Desai, N.B.; Stein, N.F.; Laquaglia, M.P.; Alektiar, K.M.; Kushner, B.H.; Modak, S.; Magnan, H.; Goodman, K.; Wolden, S. Reduced toxicity with intensity modulated radiation therapy (IMRT) for desmoplastic small round cell tumor (DSRCT): An update on the whole abdominopelvic radiation therapy (WAP-RT) experience. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, e67–e72.
- Hayes-Jordan, A.; Coakley, B.A.; Green, H.L.; Xiao, L.; Fournier, K.F.; Herzog, C.E.; Ludwig, J.A.; McAleer, M.F.; Anderson, P.M.; Huh, W.W. Desmoplastic Small Round Cell Tumor Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann. Surg. Oncol. 2018, 25, 872–877.
- Hayes-Jordan, A.; Green, H.L.; Lin, H.; Owusu-Agyemang, P.; Fitzgerald, N.; Arunkumar, R.; Mejia, R.; Okhuysen-Cawley, R.; Mauricio, R.; Fournier, K.; et al. Complete cytoreduction and HIPEC improves survival in desmoplastic small round cell tumor. Ann. Surg. Oncol. 2014, 21, 220–224.
- Campos, F.; Coutinho, D.L.; Silva, M.L.G.; Lopes, A.; Nascimento, A.; Júnior, S.A.; Nicolau, U.R.; Formiga, M.N.; Costa, F.D.; Mello, C. Clinical Characteristics, Management, and Outcomes of 19 Nonpediatric Patients with Desmoplastic Small Round Cell Tumor: A Cohort of Brazilian Patients. Sarcoma 2020, 2020, 8713165.
- Van Der Graaf, W.T.A.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886.
- Loktev, A.; Shipley, J.M. Desmoplastic small round cell tumor (DSRCT): Emerging therapeutic targets and future directions for potential therapies. Expert Opin. Ther. Targets 2020, 24, 281–285.
- Nishio, J.; Iwasaki, H.; Ishiguro, M.; Ohjimi, Y.; Fujita, C.; Yanai, F.; Nibu, K.; Mitsudome, A.; Kaneko, Y.; Kikuchi, M. Establishment and characterization of a novel human desmoplastic small round cell tumor cell line, JN-DSRCT-1. Lab. Investig. 2002, 82, 1175–1182.
- Gedminas, J.M.; Chasse, M.H.; McBrairty, M.; Beddows, I.; Kitchen-Goosen, S.M.; Grohar, P.J. Desmoplastic small round cell tumor is dependent on the EWS-WT1 transcription factor. Oncogenesis 2020, 9, 1–8.
- Van Erp, A.E.M.; Van Houdt, L.; Hillebrandt-Roeffen, M.H.S.; Van Bree, N.F.H.N.; Flucke, U.E.; Mentzel, T.; Shipley, J.; Desar, I.M.E.; Fleuren, E.D.G.; Versleijen-Jonkers, Y.M.H.; et al. Olaparib and temozolomide in desmoplastic small round cell tumors: A promising combination in vitro and in vivo. J. Cancer Res. Clin. Oncol. 2020.
- Gerald, W.L.; Rosai, J.; Ladanyi, M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc. Natl. Acad. Sci. USA 2006, 92, 1028–1032.
- Ito, E.; Honma, R.; Imai, J.-I.; Azuma, S.; Kanno, T.; Mori, S.; Yoshie, O.; Nishio, J.; Iwasaki, H.; Yoshida, K.; et al. A Tetraspanin-Family Protein, T-Cell Acute Lymphoblastic Leukemia-Associated Antigen 1, Is Induced by the Ewing’s Sarcoma-Wilms’ Tumor 1 Fusion Protein of Desmoplastic Small Round-Cell Tumor. Am. J. Pathol. 2003, 163, 2165–2172.
- Li, H.; Smolen, G.A.; Beers, L.F.; Xia, L.; Gerald, W.; Wang, J.; Haber, D.A.; Lee, S.B. Adenosine Transporter ENT4 Is a Direct Target of EWS/WT1 Translocation Product and Is Highly Expressed in Desmoplastic Small Round Cell Tumor. PLoS ONE 2008, 3, e2353.
- Kang, H.-J.; Park, J.H.; Chen, W.; Kang, S.I.; Moroz, K.; Ladanyi, M.; Lee, S.B. EWS-WT1 Oncoprotein Activates Neuronal Reprogramming Factor ASCL1 and Promotes Neural Differentiation. Cancer Res. 2014, 74, 4526–4535.
- Hingorani, P.; Dinu, V.; Zhang, X.; Lei, H.; Shern, J.F.; Park, J.; Steel, J.; Rauf, F.; Parham, D.; Gastier-Foster, J.; et al. Transcriptome analysis of desmoplastic small round cell tumors identifies actionable therapeutic targets: A report from the Children’s Oncology Group. Sci. Rep. 2020, 10, 1–12.
- Tirado, O.M.; Mateo-Lozano, S.; Notario, V. Rapamycin induces apoptosis of JN-DSRCT-1 cells by increasing the Bax: Bcl-xL ratio through concurrent mechanisms dependent and independent of its mTOR inhibitory activity. Oncogene 2005, 24, 3348–3357.
- Hayes-Jordan, A.A.; Ma, X.; Menegaz, B.A.; Lamhamedi-Cherradi, S.-E.; Kingsley, C.V.; Benson, J.A.; Camacho, P.E.; Ludwig, J.A.; Lockworth, C.R.; Garcia, G.E.; et al. Efficacy of ONC201 in Desmoplastic Small Round Cell Tumor. Neoplasia 2018, 20, 524–532.
- Shi, C.; Feng, Y.; Zhang, L.C.; Ding, D.Y.; Yan, M.Y.; Pan, L. Effective treatment of apatinib in desmoplastic small round cell tumor: A case report and literature review. BMC Cancer 2018, 18, 1–6.
- Uboldi, S.; Craparotta, I.; Colella, G.; Ronchetti, E.; Beltrame, L.; Vicario, S.; Marchini, S.; Panini, N.; Dagrada, G.P.; Bozzi, F.; et al. Mechanism of action of trabectedin in desmoplastic small round cell tumor cells. BMC Cancer 2017, 17, 107.
- Forni, C.; Minuzzo, M.; Virdis, E.; Tamborini, E.; Simone, M.; Tavecchio, M.; Erba, E.; Grosso, F.; Gronchi, A.; Aman, P.; et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol. Cancer Ther. 2009, 8, 449–457.
- Markides, C.S.; Coil, D.R.; Luong, L.H.; Mendoza, J.; Kozielski, T.; Vardeman, D.; Giovanella, B.C. Desmoplastic small round cell tumor (DSRCT) xenografts and tissue culture lines: Establishment and initial characterization. Oncol. Lett. 2013, 5, 1453–1456.
- Negri, T.; Brich, S.; Bozzi, F.; Volpi, C.V.; Gualeni, A.V.; Stacchiotti, S.; De Cecco, L.; Canevari, S.; Gloghini, A.; Pilotti, S. New transcriptional-based insights into the pathogenesis of desmoplastic small round cell tumors (DSRCTs). Oncotarget 2017, 8, 32492–32504.
- Lee, S.B.; Kolquist, K.A.; Nichols, K.; Englert, C.; Maheswaran, S.; Ladanyi, M.; Gerald, W.L.; Haber, D.A. The EWS-WT1 translocation product induces PDGFA in desmoplastic small round-cell tumour. Nat. Genet. 1997, 17, 309–313.
- Magnan, H.D.; Chou, T.; LaQuaglia, M.P.; Gerald, W.; Ladanyi, M.; Merchant, M.S. Elevated expression of VEGFR-2 and VEGFA in desmoplastic small round cell tumor (DSRCT) and activity of bevacizumab and irinotecan in a xenograft model of DSRCT. J. Clin. Oncol. 2009, 27 (Suppl. 15), 10016.
- Frezza, A.M.; Benson, C.; Judson, I.; Litière, S.; Marréaud, S.; Sleijfer, S.; Blay, J.-Y.; Dewji, R.; Fisher, C.; Van Der Graaf, W.; et al. Pazopanib in advanced desmoplastic small round cell tumours: A multi-institutional experience. Clin. Sarcoma Res. 2014, 4, 1–6.
- Bétrian, S.; Bergeron, C.; Blay, J.-Y.; Bompas, E.; Cassier, P.A.; Chevallier, L.; Fayette, J.; Girodet, M.; Guillemet, C.; Le Cesne, A.; et al. Antiangiogenic effects in patients with progressive desmoplastic small round cell tumor: Data from the French national registry dedicated to the use of off-labeled targeted therapy in sarcoma (OUTC’s). Clin. Sarcoma Res. 2017, 7, 1–7.
- Chow, L.Q.M.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 884–896.
- Menegaz, B.A.; Cuglievan, B.; Benson, J.; Camacho, P.; Lamhamedi-Cherradi, S.; Leung, C.H.; Warneke, C.L.; Huh, W.; Subbiah, V.; Benjamin, R.S.; et al. Clinical Activity of Pazopanib in Patients with Advanced Desmoplastic Small Round Cell Tumor. Oncologist 2018, 23, 360–366. Available online: https://pubmed.ncbi.nlm.nih.gov/29212731 (accessed on 6 December 2020).
- Stacchiotti, S.; Negri, T.; Zaffaroni, N.; Palassini, E.; Morosi, C.; Brich, S.; Conca, E.; Bozzi, F.; Cassinelli, G.; Gronchi, A.; et al. Sunitinib in advanced alveolar soft part sarcoma: Evidence of a direct antitumor effect. Ann. Oncol. 2011, 22, 1682–1690. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0923753419384418 (accessed on 1 September 2020).
- Italiano, A.; Kind, M.; Cioffi, A.; Maki, R.G.; Bui, B. Clinical activity of sunitinib in patients with advanced desmoplastic round cell tumor: A case series. Target Oncol. 2013, 8, 211–213.
- Scott, A.J.; Messersmith, W.A.; Jimeno, A. Apatinib: A promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs Today 2015, 51, 223. Available online: http://journals.prous.com/journals/servlet/xmlxsl/pk_journals.xml_summary_pr?p_JournalId=4&p_RefId=2320599&p_IsPs=N (accessed on 1 September 2020).
- Tian, Y.; Cheng, X.; Li, Y. Chemotherapy combined with apatinib for the treatment of desmoplastic small round cell tumors: A case report. J. Cancer Res. Ther. 2020, 16, 1177. Available online: http://www.cancerjournal.net/text.asp?2020/16/5/1177/296439 (accessed on 1 September 2020).
- Magnan, H.; Price, A.; Chou, A.J.; Riedel, E.; Wexler, L.H.; Ambati, S.R.; Slotkin, E.K.; Ulaner, G.; Modak, S.; La Quaglia, M.P.; et al. A pilot trial of irinotecan, temozolomide and bevacizumab (ITB) for treatment of newly diagnosed patients with desmoplastic small round cell tumor (DSRCT). J. Clin. Oncol. 2017, 35 (Suppl. 15), 11050.
- Fine, R.L.; Shah, S.S.; Moulton, T.A.; Yu, I.-R.; Fogelman, D.R.; Richardson, M.; Burris, H.A.; Samuels, B.L.; Assanasen, C.; Gorroochurn, P.; et al. Androgen and c-Kit receptors in desmoplastic small round cell tumors resistant to chemotherapy: Novel targets for therapy. Cancer Chemother Pharmacol. 2007, 59, 429–437.
- Bulbul, A.; Shen, J.P.; Xiu, J.; Tamayo, P.; Husain, H. Genomic and Proteomic Alterations in Desmoplastic Small Round Blue-Cell Tumors. JCO Precis Oncol. 2018, 2, 1–9.
- Thijs, A.M.J.; van der Graaf, W.T.A. Temsirolimus for Metastatic Desmoplastic Small Round Cell Tumor. Pediatr. Blood Cancer 2010, 55, 1431–1432.
- Wan, X.; Helman, L.J. The biology behind mTOR inhibition in sarcoma. Oncologist 2007, 12, 1007–1018.
- Helman, L.J.; Meltzer, P. Mechanisms of sarcoma development. Nat. Rev. Cancer 2003, 3, 685–694.
- Felkai, L.; Krencz, I.; Kiss, D.J.; Nagy, N.; Petővári, G.; Dankó, T.; Micsik, T.; Khoor, A.; Tornóczky, T.; Sápi, Z.; et al. Characterization of mTOR Activity and Metabolic Profile in Pediatric Rhabdomyosarcoma. Cancers 2020, 12, 1947.
- Subbiah, V.; Brown, R.E.; Jiang, Y.; Buryanek, J.; Hayes-Jordan, A.; Kurzrock, R.; Anderson, P.M. Morphoproteomic Profiling of the Mammalian Target of Rapamycin (mTOR) Signaling Pathway in Desmoplastic Small Round Cell Tumor (EWS/WT1), Ewing’s Sarcoma (EWS/FLI1) and Wilms’ Tumor(WT1). PLoS ONE 2013, 8, e68985.
- Jiang, Y.; Subbiah, V.; Janku, F.; Ludwig, J.A.; Naing, A.; Benjamin, R.S.; Brown, R.E.; Anderson, P.; Kurzrock, R. Novel secondary somatic mutations in Ewing’s sarcoma and desmoplastic small round cell tumors. PLoS ONE 2014, 9, e93676.
- Tarek, N.; Hayes-Jordan, A.; Salvador, L.; McAleer, M.F.; Herzog, C.E.; Huh, W.W. Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr. Blood Cancer 2018, 65, 1–3.
- Naing, A.; Lorusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mtor inhibitor temsirolimus in patients with refractory ewing’s sarcoma family tumors. Clin. Cancer Res. 2012, 18, 2625–2631.
- Katz, D.; Azraq, Y.; Eleyan, F.; Gill, S.; Peretz, T.; Merimsky, O. Pazolimus: Pazopanib plus sirolimus following progression on pazopanib, a retrospective case series analysis. BMC Cancer 2016, 16, 616.

