 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Serif Senturk | + 3853 word(s) | 3853 | 2021-01-28 10:43:10 | | | |
| 2 | Rita Xu | -1888 word(s) | 1965 | 2021-02-04 09:44:44 | | |
Video Upload Options
Cellular senescence is a state of stable cell cycle arrest that can be triggered in response to various insults and is characterized by distinct morphological hallmarks, gene expression profiles, and the senescence-associated secretory phenotype (SASP). Importantly, cellular senescence is a key component of normal physiology with tumor suppressive functions.
1. Introduction
Almost 60 years ago, Hayflick and Moorhead challenged Carrel’s original proposition that normal cells have an infinite replication capacity. On that account, Hayflick performed a series of experiments with diploid primary cells derived from various human embryonic tissues. These studies unveiled the fact that normal cells propagated in culture can replicate for a limited and probably predetermined number of generations, after which they undergo an irreversible arrest of cell growth, thus disproving Carrel’s theory of cellular immortality [1][2]. This phenomenon is now known as the Hayflick limit or, as it will be called herein, replicative senescence [3][4][5][6]. Over the course of six decades, cellular senescence has been established as an adaptive stress response mechanism in physiological and pathological processes with both beneficial and detrimental consequences for human health [7][8][9][10].
Depending on the cell type and conditions, different subtypes of cellular senescence such as DNA-damage-induced senescence, stress-induced senescence (SIS), and oncogene-induced senescence (OIS) have been defined [11][12][13]. Earlier studies have shown that cellular senescence program is a key component of embryonic development and tissue remodeling and may potentially function as a tumor suppressor mechanism against carcinogenesis [9][14][15]. Work in recent decades have debated the longstanding fundamental paradigm of senescence irreversibility. In striking contrast to the traditional definition, these research efforts have provided mounting evidence that this complex phenotype is not a static, permanent, and docile state, but rather entails a constantly evolving multi-step process with cell-autonomous and non-cell-autonomous implications and often deleterious effects on tissue homeostasis. In the context of cancer therapy, this capacity is primarily due to the fact that senescent cells remain viable and bioactive for long periods of time and eventually resume proliferation while emitting heterotypic signals to their microenvironment [16][17][18][19].
The century-old classic novel “The Strange Case of Dr. Jekyll and Mr. Hyde” by Robert Louis Stevenson explores the duality of human nature—specifically, the natural existence of a dual personality, good and evil, in the same individual. Arguably, the balance between good and evil is what makes us human. In some sense, this theme is analogous to the dual nature of cellular senescence, where it can be both beneficial (Jekyll) and detrimental (Hyde). Our objective in this review is to synthesize the recent scientific advances pertaining to the duality of cellular senescence, with a heightened interest in cancer, and to present scientific advances and challenges in exploiting this phenotype in cancer therapies. With this motivation, we first revisit the hallmarks of cellular senescence primarily by stressing the morphological and molecular biomarkers, as well as the regulation and functions of the senescence-associated secretory phenotype (SASP). The key effector mechanisms and different subtypes of cellular senescence are then briefly summarized. Next, we discuss the biological significance of cellular senescence in normal physiology and shift the focus to cancer, referencing the good and evil natures of this phenomenon.
2. The Hallmarks and Molecular Mechanisms of Cellular Senescence
2.1. Morphological and Molecular Biomarkers of Senescent Cells
Typically characterized by the inability to replicate their DNA and cellular growth arrest, cultured senescent cells exhibit a series of distinct morphological and chemical hallmarks which distinguish them from proliferating cells. Perhaps the most notable molecular markers are multiple or enlarged nuclei and flattened cytoplasm, an increased number of lysosomes and Golgi apparatus, elevated pH-dependent senescence-associated b-galactosidase activity (SA-b-gal), and resistance to apoptosis [16][20][21]. Senescent cells are also frequently characterized by impaired nuclear integrity; the formation of persistent nuclear DNA damage foci and DNA-damage response (DDR); deregulated metabolism; protein and lipid damage; global epigenetic changes in their chromatin landscape; the formation of senescence-associated heterochromatin foci (SAHF); and, of course telomere attrition, the hallmark of replicative senescence [12][13][22][23][24] (Figure 1).
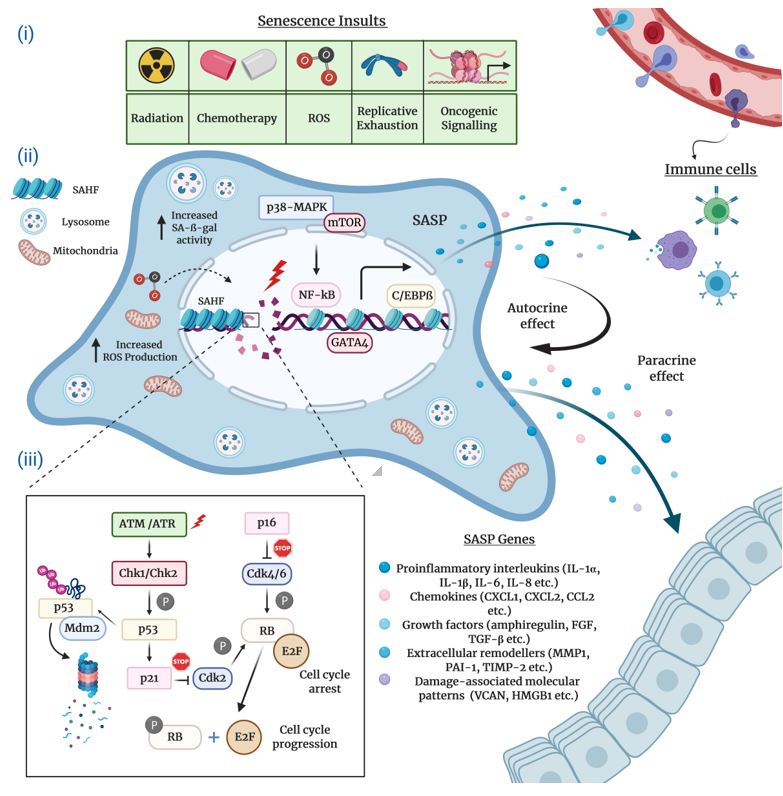
Figure 1. The hallmarks and molecular mechanisms of cellular senescence. The figure summarizes 3 major attributes of cellular senescence. (i) Intrinsic and extrinsic insults causing senescence: The extrinsic factors causing senescence-related cell cycle arrest comprise of radiation and chemotherapy, whereas intrinsic factors harbor increased reactive oxygen species (ROS) accumulation, aberrant oncogene activation, and replicative exhaustion. (ii) Molecular hallmarks of senescent cells and pathways regulating senescence-associated secretory phenotype (SASP) production and immune cell infiltration (recruitment of immune cells to the SASP-rich milieu): SASP expression is predominantly controlled by the p38-MAPK and mTOR pathways and C/EBPβ, GATA4, NF-κB transcription factors. Senescent cells enriched for SASPs disseminate a wide assortment of senescence cues (proinflammatory interleukins, chemokines, growth factors, extracellular remodelers, damage-associated molecular patterns/DAMPs) to the surrounding cells (paracrine effect). At the same time, these cues influence on the senescent cell itself (autocrine effect). Clearance of senescent cells is actualized via immune surveillance mechanisms, and (iii) molecular mechanisms modulating cell cycle arrest: DNA-damage dependent and DNA-damage independent mechanisms regulate the key effector mechanisms p53/p21Cip1 and pRb/p16Ink4a to initiate and maintain cellular senescence.
2.2. The Senescence-Associated Secretory Phenotype (SASP) of Senescent Cells
A striking feature of virtually all senescent cells is the widespread changes in protein expression that involve a specific signature for secreted molecules, collectively known as the SASP. The SASP consists of a myriad of biologically active soluble and insoluble factors which can be grouped into the following major categories: proinflammatory interleukins and chemokines; growth factors; extracellular matrix proteins and remodeling enzymes; damage-associated molecular patterns; and extracellular vehicles greatly enriched for enzymes, miRNAs, and DNA fragments [25][26][27][28][29][30]. Recently, Basisty et al. developed a comprehensive and quantitative proteomic atlas that can potentially serve as a reference and guide for the identification of novel soluble (sSASP) and exosome/extracellular vesicle SASP (eSASP) factors. The atlas is currently limited to two distinct cell lines induced to senesce by various stress factors. However, the authors expect the resource to be continuously updated by depositing new SASP profiles derived from different cell types and senescence-inducing conditions [30]. In essence, the abundance and heterogeneous composition of the SASP is context-dependent, partly explaining how the SASP can exert profoundly diverse and sometimes contradictory functions in numerous biological processes such as tissue remodeling, inflammation, and age-related pathologies including cancer [31].
The SASP regulation in senescent cells has been the subject of numerous studies. The findings collected in these studies substantiate the notion that the SASP production is coordinated by a complex network of signaling cascades that involve to a large extent transcriptional but also post-transcriptional mechanisms (Figure 1). Stress-inducible kinase p38 mitogen-activated protein kinase (p38-MAPK), mammalian target of rapamycin (mTOR), cytosolic DNA-sensing cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING), the Ataxia telangiectasia mutated (ATM)/ATM- and RAD-3 related (ATR)-activated IκB kinase (IKK)/NEMO complex, and the GATA binding protein 4 (GATA4) axis constitute the most prominent upstream regulators of the pro-inflammatory senescence phenotype [31][32][33][34][35][36]. Upon stimulation by stress conditions, these interactive signaling pathways converge towards the activation of a transcriptional program managed by the nuclear factor kappa B (NF-κB) and the CCAAT-enhancer binding protein b (C/EBPb), the core effectors that initiate and maintain SASP gene expression [31][37][38]. A number of studies suggest that the Janus kinase–signal transducer and activator of transcription (JAK/STAT) and NOTCH pathways also play a crucial role in the transcriptional regulation of SASP components through C/EBPb [39][40][41].
The DDR signaling pathway is a critical mediator of the SASP. Available research indicates that the direct activation of ATM/ATR protein kinases in response to persistent DNA damage inhibits the autophagic degradation of GATA4, which, in turn, activates NF-κB to initiate and maintain the SASP network [42][43]. Differently, several reports describe the DNA damage-independent control of the SASP induction, which, in general, involves the p38-MAPK-mediated activation of NF-κB [32][44][45][46][47]. Intriguingly, mitochondrial dysfunction-associated senescence (MiDAS) is a distinct form of DDR-independent cellular senescence wherein the cells undergoing MiDAS display a unique SASP profile dictated by AMP-activated protein kinase (AMPK)-mediated p53 activation [47].
Epigenetic mechanisms are also pronounced in the modulation of cellular senescence and SASP constituents. For example, the histone variants macroH2A1 and H2AJ accumulate in human primary lung fibroblasts during OIS and play an important role in the positive and negative regulation of SASP production [48][49]. Similarly, epigenetic modifiers including lysine methyltransferase 2A (KMT2A, also known as MLL1), high-mobility group B protein 1 and 2 (HMGB1 and HMGB2), and bromodomain-containing protein 4 (BRD4) modulate the senescence secretome by orchestrating the chromatin landscape around the SASP gene loci [50][51][52][53]. Moreover, the downregulation of sirtuin 1 (SIRT1) gene and the enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) gene in senescent cells positively regulate SASP factors, which are mediated by transcriptomic changes through the post-translational modifications of histones [46][54]. In addition to transcriptional mechanisms, the expression of SASP genes is regulated at the post-transcriptional level. In particular, MAPK-activated protein kinase 2 (MK2), a downstream effector of the p38-MAPK and mTOR pathways, modulates the mRNA stability of a subset of SASP components by means of ARE-mediated decay [55][56].
2.3. Molecular Mechanisms Underlying Cellular Senescence
The complex network of molecular events that execute cellular senescence has been extensively reviewed elsewhere [40][57][58]. Nonetheless, for the sake of the completeness and consistency of this review, we will mention the critical effector pathways. Many lines of research convincingly attest that the onset and maintenance of permanent senescence arrest is controlled by the p53/p21Cip1 and the retinoblastoma protein (pRb)/p16Ink4a tumor suppressor pathways (Figure 1). In principle, the activation of either one or both of these crucial pathways can readily induce cellular senescence. Notably, genetic mutations or epigenetic silencing of these pathways obliterates the senescence response in most cell types, occasionally paving the way for cancer initiation and progression [59].
Mechanistically, in its active hypophosphorylated form the pRb binds to and sequesters E2F family of transcription factors and induces growth arrest in the G1 phase of the cell cycle. To achieve this, pRb suppresses the transcription of several E2F target genes encoding a repertoire of essential proteins indispensable for DNA replication and cell cycle, thus blocking the subsequent entry into and progression through the S phase. The regulation of cellular senescence by E2F is often correlated with context-dependent local or global structural epigenetic modifications such as chromatin remodeling and SAHF formation. Consistent with this, the promoters of E2F target genes are enriched for repressive histone modifications (mainly H3K9me3 and H3K27me3) which result in gene expression changes that eventually contribute to the regulation of cellular senescence. Upon phosphorylation by Cyclin D and Cyclin-dependent kinase 4 and 6 (CDK4 and CDK6), a complex that is negatively regulated by the p16Ink4a tumor suppressor protein, pRb switches to an inactive state and releases E2F, thus stimulating cell cycle progression [60][61][62][63].
The p53 transcription factor, the guardian of the genome integrity, plays a pivotal role in the induction and maintenance of cellular senescence. Following exposure to genotoxic or non-genotoxic stress, p53 gets activated and promotes cell cycle arrest via DDR-dependent and DDR-independent mechanisms. The specific activity of p53 is tightly controlled by virtue of positive and negative regulators and post-translational modifications. In response to stress stimuli, p53 is phosphorylated and stabilized by ATM/ATR and Checkpoint kinase 1 and 2 (Chk1/2) protein kinases, releasing it from MDM2, an E3 ubiquitin ligase that negatively regulates p53 via ubiquitination and proteasomal degradation. Once activated, p53 selectively increases the transcription of various target genes, in particular p21Cip1 (CDKN1A), a potent CDK inhibitor which executes the p53-mediated control of cellular senescence. The p21Cip1 protein binds to and inhibits the activity of Cyclin E/CDK2 and Cyclin D/CDK4 complexes, thus activating pRb and blocking cell cycle progression [17][64][65][66]. Finally, yet importantly, depending on the cellular identity and stress factors, antitumor mechanisms coordinated by the p53 and pRb pathways may engage different subtypes of cellular senescence.
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Cell Res. 1961, 25, 585–621, doi:10.1016/0014-4827(61)90192-6.
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Cell Res. 1965, 37, 614–636, doi:10.1016/0014-4827(65)90211-9.
- Campisi, J. Replicative Senescence: An Old Lives’ Tale? Cell 1996, 84, 497–500, doi:10.1016/S0092-8674(00)81023-5.
- Shay, J.W.; Wright, W.E. Hayflick, His Limit, and Cellular Ageing. Rev. Mol. Cell Biol. 2000, 1, 72–76, doi:10.1038/35036093.
- Fagagna, F. d’Adda di; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198, doi:10.1038/nature02118.
- Goldstein, S. Replicative Senescence: The Human Fibroblast Comes of Age. Science 1990, 249, 1129–1133, doi:10.1126/science.2204114.
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking Cellular Stress Responses to Systemic Homeostasis. Rev. Mol. Cell Biol. 2018, 19, 731–745, doi:10.1038/s41580-018-0068-0.
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Rev. Mol. Cell Biol. 2007, 8, 729–740, doi:10.1038/nrm2233.
- Van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446, doi:10.1038/nature13193.
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. Cell Biol. 2018, 217, 65–77, doi:10.1083/jcb.201708092.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Med. 2015, 21, 1424–1435, doi:10.1038/nm.4000.
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Rev. Mol. Cell Biol. 2014, 15, 482–496, doi:10.1038/nrm3823.
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The Essence of Senescence. Genes Dev. 2010, 24, 2463–2479, doi:10.1101/gad.1971610.
- Narita, M.; Lowe, S.W. Senescence Comes of Age. Med. 2005, 11, 920–922, doi:10.1038/nm0905-920.
- Campisi, J. Cellular Senescence as a Tumor-Suppressor Mechanism. Trends Cell Biol. 2001, 11, S27–S31, doi:10.1016/s0962-8924(01)02151-1.
- Lozano-Torres, B.; Estepa-Fernández, A.; Rovira, M.; Orzáez, M.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. The Chemistry of Senescence. Rev. Chem. 2019, 3, 426–441, doi:10.1038/s41570-019-0108-0.
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J. 2003, 22, 4212–4222, doi:10.1093/emboj/cdg417.
- Hoare, M.; Narita, M. Transmitting Senescence to the Cell Neighbourhood. Cell Biol. 2013, 15, 887–889, doi:10.1038/ncb2811.
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The Role of Senescence in Cancer Development. Cancer Biol. 2020, 62, 182–191, doi:10.1016/j.semcancer.2019.06.018.
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Natl. Acad. Sci. USA 1995, 92, 9363–9367, doi:10.1073/pnas.92.20.9363.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453, doi:10.1016/j.tcb.2018.02.001.
- Zhang, R.; Chen, W.; Adams, P.D. Molecular Dissection of Formation of Senescence-Associated Heterochromatin Foci. Cell. Biol. 2007, 27, 2343–2358, doi:10.1128/MCB.02019-06.
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839, doi:10.1016/j.cell.2016.07.050.
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence-Size Matters Not. EBioMedicine 2017, 21, 14–20, doi:10.1016/j.ebiom.2017.03.027.
- Kuilman, T.; Peeper, D.S. Senescence-Messaging Secretome: SMS-Ing Cellular Stress. Rev. Cancer 2009, 9, 81–94, doi:10.1038/nrc2560.
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Cell Biol. 2013, 15, 978–990, doi:10.1038/ncb2784.
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. Clin. Invest. 2013, 123, 966–972, doi:10.1172/JCI64098.
- Wallis, R.; Mizen, H.; Bishop, C.L. The Bright and Dark Side of Extracellular Vesicles in the Senescence-Associated Secretory Phenotype. Ageing Dev. 2020, 189, 111263, doi:10.1016/j.mad.2020.111263.
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small Extracellular Vesicles and Their MiRNA Cargo Are Anti-Apoptotic Members of the Senescence-Associated Secretory Phenotype. Aging 2018, 10, 1103–1132, doi:10.18632/aging.101452.
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biol. 2020, 18, e3000599, doi:10.1371/journal.pbio.3000599.
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Rev. Cancer 2019, 19, 439–453, doi:10.1038/s41568-019-0156-2.
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK Is a Novel DNA Damage Response-Independent Regulator of the Senescence-Associated Secretory Phenotype. EMBO J. 2011, 30, 1536–1548, doi:10.1038/emboj.2011.69.
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev. 2020, 34, 1565–1576, doi:10.1101/gad.343129.120.
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The Senescence-Associated Secretory Phenotype and Its Regulation. Cytokine 2019, 117, 15–22, doi:10.1016/j.cyto.2019.01.013.
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through CGAS Promotes Senescence. Cell Biol. 2017, 19, 1061–1070, doi:10.1038/ncb3586.
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406, doi:10.1038/nature24050.
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136, doi:10.1101/gad.17276711.
- Salotti, J.; Johnson, P.F. Regulation of Senescence and the SASP by the Transcription Factor C/EBPβ. Gerontol. 2019, 128, 110752, doi:10.1016/j.exger.2019.110752.
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018, doi:10.1016/j.cell.2008.03.038.
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Cell Biol. 2019, 21, 94–101, doi:10.1038/s41556-018-0249-2.
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 Mediates a Switch between Two Distinct Secretomes during Senescence. Cell Biol. 2016, 18, 979–992, doi:10.1038/ncb3397.
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Cell Biol. 2009, 11, 973–979, doi:10.1038/ncb1909.
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612, doi:10.1126/science.aaa5612.
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A Key Role for Mitochondrial Gatekeeper Pyruvate Dehydrogenase in Oncogene-Induced Senescence. Nature 2013, 498, 109–112, doi:10.1038/nature12154.
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118, doi:10.1016/j.cell.2013.10.019.
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492, doi:10.1016/j.celrep.2018.03.002.
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314, doi:10.1016/j.cmet.2015.11.011.
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Cell 2015, 59, 719–731, doi:10.1016/j.molcel.2015.07.011.
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone Variant H2A.J Accumulates in Senescent Cells and Promotes Inflammatory Gene Expression. Commun. 2017, 8, 14995, doi:10.1038/ncomms14995.
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 Is a Central Mediator of Senescent Phenotypes. Cell Biol. 2013, 201, 613–629, doi:10.1083/jcb.201206006.
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261, doi:10.1158/0008-5472.CAN-11-3386.
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629, doi:10.1158/2159-8290.CD-16-0217.
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 Is Essential for the Senescence-Associated Secretory Phenotype. Genes Dev. 2016, 30, 321–336, doi:10.1101/gad.271882.115.
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS ONE 2015, 10, e0116480, doi:10.1371/journal.pone.0116480.
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. P38MAPK Plays a Crucial Role in Stromal-Mediated Tumorigenesis. Cancer Discov. 2014, 4, 716–729, doi:10.1158/2159-8290.CD-13-0743.
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. MTOR Regulates MAPKAPK2 Translation to Control the Senescence-Associated Secretory Phenotype. Cell Biol. 2015, 17, 1205–1217, doi:10.1038/ncb3225.
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting Senescent Cells in Translational Medicine. EMBO Mol. Med. 2019, 11, e10234, doi:10.15252/emmm.201810234.
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. Clin. Invest. 2018, 128, 1238–1246, doi:10.1172/JCI95148.
- Sherr, C.J.; McCormick, F. The RB and P53 Pathways in Cancer. Cancer Cell 2002, 2, 103–112, doi:10.1016/S1535-6108(02)00102-2.
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827, doi:10.1016/j.cell.2019.10.005.
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716, doi:10.1016/s0092-8674(03)00401-x.
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387, doi:10.1016/j.ccr.2010.01.023.
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of P16CDKN2 Expression and Its Implications for Cell Immortalization and Senescence. Cell. Biol. 1996, 16, 859–867, doi:10.1128/mcb.16.3.859.
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial Role of P53-Dependent Cellular Senescence in Suppression of Pten-Deficient Tumorigenesis. Nature 2005, 436, 725–730, doi:10.1038/nature03918.
- Itahana, K.; Dimri, G.; Campisi, J. Regulation of Cellular Senescence by P53: P53 and Cellular Senescence. J. Biochem. 2001, 268, 2784–2791, doi:10.1046/j.1432-1327.2001.02228.x.
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and Aging: The Critical Roles of P53. Oncogene 2013, 32, 5129–5143, doi:10.1038/onc.2012.640.


