 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Perez-Zsolt | + 4979 word(s) | 4979 | 2021-01-29 06:52:17 | | | |
| 2 | Dean Liu | -1951 word(s) | 3028 | 2021-02-04 08:03:00 | | |
Video Upload Options
Ebola virus (EBOV), which belongs to the filoviridae family, and the recently emerged coronavirus SARS-CoV-2 are two highly pathogenic viruses that exploit very similar endocytic routes to productively infect target cells. This convergence has sped up the experimental assessment of clinical therapies against SARS-CoV-2 previously found to be effective for EBOV, and facilitated their expedited clinical testing.
1. Introduction
SARS-CoV-2 was identified on 7 January, 2020 as the etiological agent responsible for COVID-19, a severe respiratory disease currently causing a global pandemic. Since then, research groups worldwide have dedicated their efforts to understand the viral cycle of this new coronavirus and to find strategies to prevent infection. As of 12 December 2020, there were almost 70 million cases confirmed and more than 1.5 million deaths affecting 220 countries in the globe, sparkling global concern (https://www.who.int/emergencies/diseases/novel-coronavirus-2019?gclid=Cj0KCQiA8dH-BRD_ARIsAC24umbD-JsU2gwShKk7Q6H1RJ-lo0JZuRG8to08SFLhF6BL1YuRf4I-lHYaAn9aEALw_wcB). The high transmissibility of the virus, the broad range of symptoms associated to the disease and the lack of effective therapeutics to prevent the course of the infection has sped up the search for novel treatments and vaccines.
A similar challenge was faced by the scientific community between 2013 and 2020, when Ebola virus (EBOV) threatened humankind causing two major outbreaks in Central and West Africa, which caused an Ebola Virus Disease (EVD) that presented up to a 90% case-fatality rate. The incredible amount of scientific knowledge generated during the EBOV epidemic identified antivirals displaying efficacy against different steps in the EBOV life cycle, therapeutic neutralizing antibodies, and vaccine strategies. All these tools laid the foundations to better cope with future viral zoonotic infections. Some of these strategies have also been deployed against SARS-CoV-2, and the early efficacy shown in vitro has demonstrated key similitudes between both zoonotic viruses.
2. Setting the Stage for Infection: Viral Binding and Host Attachment Receptors for EBOV and SARS-CoV-2
The very first step of the viral life cycle is the attachment of the virus via key receptors, followed by a viral entry process that relies on the same or alternative host factors that finally lead to productive infection. The availability of these critical host attachment molecules determines the tissue tropism, which greatly varies depending on the type of virus. Since the specific steps of viral binding and subsequent entry are shared among very distant viruses, lessons learned in the past can illuminate how a new virus like SARS-CoV-2 interacts with target cells. The spectrum of cellular molecules that act as virus attachment receptors is extremely broad, and viruses mostly can bind to more than one factor on the host cell membrane (Figure 1). Such is the case of EBOV, whose affinity to a wide variety of host cell receptors mediates viral binding to different cellular targets (Figure 1A). C-type lectins (CLECs), which are able to interact with particular glycans exposed on the viral glycoproteins, comprise DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), L-SIGN (liver/lymph node-specific intercellular adhesion molecule-3-grabbing non-integrin), hMGL (human macrophage galactose- and N-acetylgalactosamine-specific C-type lectin), and mannose-binding lectins, all of which bind to N- and O-linked glycans on Ebola virus glycoprotein, as reviewed in[1]. However, cells lacking CLEC expression remain permissive for EBOV infection. Importantly, phosphatidyl serine (PtdSer) binding receptors can also recognize this lipid exposed on the viral envelope of EBOV. PtdSer-recognizing receptors include protein complexes composed of Gas6 or protein S, members of the T-cell immunoglobulin and mucin domain (TIM) family TIM-1 and TIM-4, and the TAM family of receptor tyrosine kinases Tyro3, Axl, and Mer[2] (Figure 1A). EBOV binding efficiency also depends on the presence of plasma membrane sphingomyelin, and the activity of acid sphingomyelinase (ASMase)[3]. In activated myeloid cells, EBOV entry is enhanced by the sialic acid-binding Ig-like lectin 1 (Siglec-1/CD169), which recognizes sialylated gangliosides exposed on the cellular-derived membrane of the virus[4] (Figure 1A). Overall, these cellular receptors contribute to EBOV attachment and promote subsequent infection.
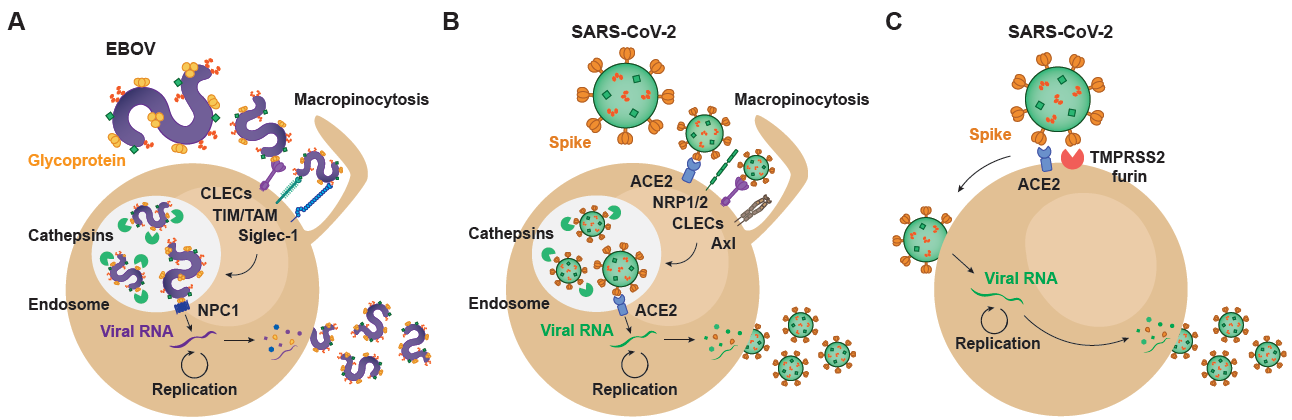
Figure 1. Viral and host factors involved in EBOV and SARS-CoV-2 infectious cycle. (A) EBOV entry into target cells is mediated by macropinocytosis, which directs surface-attached viral particles to the endosomal trafficking pathway. Within endosomes, host cathepsins cleave viral glycoprotein, facilitating interaction with the NPC1 receptor and viral membrane fusion. In the cytoplasm, the viral RNA genome undergoes transcription/replication, resulting in the synthesis of new viral particles that exit infected cells through membrane budding. (B) SARS-CoV-2 can enter target cells through an endosomal pathway that parallels EBOV internalization. Within endosomal compartments, cleavage of the Spike protein results in viral fusion and cytoplasmic entry, where viral replication occurs. (C) SARS-CoV-2 also enters target cells through an alternative mechanism in which Spike protein is cleaved at the cell surface, a process mediated by proteases such as TMPRSS2 and furin. In this case, the viral genome gains access to the cytoplasm through viral fusion with the plasma membrane. EBOV: Ebola virus; CLECs: C-type lectin receptors; TIM: T-cell immunoglobulin and mucin receptors; TAM: Tyro3-Axl-Mer receptors; Siglec-1: sialic acid-binding Ig-like lectin 1; NPC1: Niemann-Pick receptor C1; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; ACE2: angiotensin-converting enzyme 2; NRP1/2: neuropilin 1/2; TMPRSS2: transmembrane protease serine 2.
On the contrary, SARS-CoV-2 attachment to susceptible cells remains primarily on the binding of the Spike protein to the host angiotensin converting enzyme 2 (ACE2) on the susceptible cell membrane[5] (Figure 1B,C), and most importantly, ACE2 is also critical for viral fusion[6]. However, other factors may also actively interact with SARS-CoV-2 and promote viral binding and attachment. Clausen and colleagues demonstrated that heparan sulfate, which is a highly negatively charged polysaccharide attached to proteoglycans found on the cellular membrane or the extracellular matrix, interacts with the ectodomain of the SARS-CoV-2 Spike protein to shift its conformation and allow binding to ACE2[7]. Integrins are also proposed as potential players in the entry of SARS-CoV-2 into the host cell[8], and the Spike protein has a specific motif able to bind these receptors. Integrin alfa and beta molecules recognize specific motifs in the spike protein of SARS-CoV-2 and have the potential to trigger infection by binding integrin heterodimers, activating transducing pathways involving phosphatidylinositol-3 kinase (PI-3K) or mitogen-activated protein kinase (MAPK), which can promote viral entry[8]. Thus, as already reported for EBOV, binding to integrins can facilitate SARS-CoV-2 endocytosis and infection. Neuropilin-1 and 2 (NRP1 and NRP2) have also recently been reported to play a role on SARS-CoV-2 attachment[9][10] (Figure 1B). Although the absence of these proteins still allows for viral entry into susceptible cells, infectivity gets reduced. Daly et al. hypothesize that the upregulation of neuropilins in lung tissues of COVID-19 patients and their binding to the SARS-CoV-2 spike protein may be one of the reasons to explain why this virus is more infectious than SARS-CoV-1[10]. Another potential receptor for SARS-CoV-2 is the CD147 or extracellular matrix metalloproteinase inducer (EMMPRIN), a protein that belongs to the immunoglobulin superfamily enrolled in inflammatory processes and viral cellular entry[11].
Many of the early events that govern attachment of SARS-CoV-2 to cellular targets remain still unknown and require further investigation. CLECs already implicated in EBOV binding, such as DC-SIGN and L-SIGN, have also been associated with the capacity to transmit SARS-CoV-2 pseudoviruses to target cells expressing ACE2[12] (Figure 1A,B). Moreover, SARS-CoV-2 specifically interacts with tyrosine-protein kinase receptor UFO (Axl) on the host membrane, where this receptor can promote viral entry[13], as already described for EBOV (Figure 1A,B). Interestingly and also previously reported for EBOV, fluoxetine, a functional inhibitor of ASMase, efficiently abrogates the SARS-CoV-2 entry and propagation in Vero E6 and CaLu-3 cells, suggesting that ASMase may also play a significant role in the early steps of the virus infection cycle[3][14]. A further understanding of the role of attachment factors implicated in SARS-CoV-2 binding will be required to reduce systemic dissemination between susceptible cells and tissues. Moreover, studying how these attachment factors set up the stage and facilitate viral entry and fusion will be critical to develop effective antiviral strategies.
3. EBOV Entry Converges with the Endosomal Route of SARS-CoV-2
Distinct viruses have evolved to use endocytic pathways to promote efficient infection, which requires the delivery of the viral genome into the cell cytoplasm at sites where replication proceeds optimally. Both SARS-CoV-2 and EBOV can utilize analogous pH-dependent endocytic routes to enter the cytoplasm of infected cells, since their viral proteins rely on similar proteolytic cleavage mechanisms that can take place at endosomal compartments. In the particular case of SARS-CoV-2 though, alternative entry processes at the plasma membrane are also key determinants of the pathogenesis of this coronavirus, as we will later discuss.
SARS-CoV-2 viral entry is mediated by the interaction of the Spike viral protein with ACE2, that allows for viral fusion and infection[6]. The Spike protein is comprised of two major units. The N-terminal S1 subunit contains the receptor binding domain (RBD), which is essential for attachment to ACE2. The C-terminal S2 subunit harbors key domains that play a role in membrane fusion and intracellular trafficking into the cytoplasm[15]. As reported in previous coronavirus studies, the cleavage of the Spike protein at the boundary between the S1 and S2 subunits by cellular host proteases is required for the activation of the protein to promote virus–cell fusion[16][17]. Indeed, there is an additional furin-type cleavage site at the junction between S1 and S2 of the newly discovered coronavirus that was not originally present in SARS-CoV-1, and is assumed to comparatively enhance SARS-CoV-2 infectivity[9][19]. Following the cleavage by furin of the S protein, the RBD of the S1 subunit of SARS-CoV-2 binds to the outer surface of ACE2 with a higher affinity compared to SARS-CoV-1 RBD[19][20]. This engagement triggers a conformational rearrangement that causes S1 shedding, cleavage of the S2 subunit by host proteases and exposure of a fusion peptide located next to the proteolytic side in S2 [17][21][22].
While the novel coronavirus mainly fuses at the cellular membrane of susceptible cells, where particular host proteases with the capacity to prime the Spike protein such as TMPRSS2 or TMPRSS4 are exposed, this virus can also exploit an alternative endocytic route[23] (Figure 1B). In certain cellular types, SARS-CoV-2 can also enter the cells via intracellular endosomal compartments, where other host proteases such as cathepsins can prime the Spike and promote viral fusion with internal endosome membranes[6]. This later endocytic route clearly resembles to that followed by EBOV, which is also internalized through an endosomal pathway that triggers viral fusion (Figure 1A,B). Following virus–cell attachment, EBOV is internalized primarily by macropinocytosis[24] (Figure 1A). Although other routes of uptake have been reported, including caveolin- and clathrin-dependent endocytosis, many of those studies have been performed with retroviral pseudotypes, which in the case of EBOV, do not display native virus morphology nor viral glycoprotein density and other biochemical characteristics[25].
As it happens with the Spike protein of SARS-CoV-2, EBOV contains a viral glycoprotein at the outer surface that mediates virus and host membranes fusion upon cellular protease cleavage. The mature conformations of GP with capacity to fuse with endosomal membranes requires a post-translational furin cleavage. This process produces a disulfide heterodimer composed of GP1 and GP2 subunits, being the former required for receptor interactions and the latter required for membrane fusion[26]. After initial internalization, virus particles are trafficked to the late endosomes/lysosomes through the endo-lysosomal pathway, where pH decreases and cysteine proteases cathepsins B/L cleave EBOV GP1 into its fusogenic form, which has the RBD exposed[27][28][29] (Figure 1A). Cathepsins L and B where initially identified as the essential proteases for the processing of EBOV GP and, indeed, their cleavage sites within the viral glycoprotein sequence have been mapped. The processed GP1 interacts with the late endosomal/lysosomal Niemann-Pick C1 (NPC1) intracellular receptor, which triggers the fusion of the viral envelope with the cellular endosomal membrane upon GP2 dependency[30][31] (Figure 1A). Although the specific mechanism is still not clear, the membrane fusion step also requires the activity of the Two-Pore Calcium Channel 2 (TPC2) in the endosomal membrane[32].
In the case of SARS-CoV-2, this endosomal viral entry pathway requires the binding of the Spike protein to ACE2 and its priming by cathepsin proteases[33] (Figure 1B). Thus, the cathepsin-mediated cleavage is a critical step for the entry of SARS-CoV-2 and EBOV. It is important to remember, however, that in contrast to EBOV, which can only fuse in endocytic compartments, SARS-CoV-2 mainly exploits the plasma membrane for accessing cellular targets in which specific serine proteases are able to prime the Spike of the coronavirus at the plasma membrane (Figure 1C). Proteolytic cleavage of the Spike protein by TMPRSS2 allows fusion at the plasma membrane of key cellular targets. As we will later discuss, this complicates the clinical use of cathepsin inhibitors and therapeutic agents that interfere with the endocytic route of entry for SARS-CoV-2, which displays an independent viral fusion pathway at the plasma membrane that is highly active in pulmonary cells[6]. Studying cellular gateways exploited by very distant viruses may aid to identify hot spots where viral entry converges, what will be key to develop broad pan-antiviral strategies aimed at avoiding infection. Once viral fusion takes place, productive infection will trigger viral replication and complicate viral control.
4. Transcription and Replication of EBOV and SARS-CoV-2
Once EBOV and SARS-CoV-2 genomes are released into the cell cytoplasm, viral replication occurs through a tightly regulated process involving viral and host factors (Figure 1). As both EBOV and SARS-CoV-2 are single-stranded RNA viruses, they share common features in their transcription and replication processes. However, the opposite polarity of their genomes also implicates the existence of relevant divergences between them. In this section, we will analyze the differences and similarities for EBOV and SARS-CoV-2 transcription and replication.
EBOV negative-sense RNA genome enters the cytoplasm in the form of a ribonucleoprotein complex. Viral genome is encapsidated by EBOV nucleoprotein (NP), and it is associated to the RNA-dependent RNA polymerase (L) and viral proteins 35 (VP35), 30 (VP30), and 24 (VP24), which play critical roles in viral transcription and replication. VP24 mediates viral uncoating, making the genome accessible to the transcription machinery[34][35]. VP35 and VP30 serve as co-factors for the L polymerase, that generates positive-sense mRNAs encoding the viral proteins using the viral genome as a template[36]. Following this primary transcription process, secondary transcription cycles are mediated by the newly synthesized viral polymerase and co-factors, thus amplifying the production and accumulation of cytoplasmic viral proteins[36] (Figure 1A).
In contrast to EBOV, coronaviruses have a positive-sense RNA genome that is readily translated by the host machinery upon cytoplasmic entry (Figure 1B,C). Translation of SARS-CoV-2 open reading frame 1a (ORF1a) and 1ab (ORF1b) results in the synthesis of the polyproteins 1a (pp1a) an 1ab (pp1ab), respectively[37]. These polyproteins need further processing to give rise to functional non-structural proteins 1-16 (nsp1-16), which contribute to the formation of a replication complex observed in other coronavirus species[38][39][40]. Moreover, they facilitate the synthesis of viral proteins by inhibiting the translation of host proteins[41][42]. Among non-structural proteins, the major protease nsp5 (Mpro) and the papain-like protease nsp3 (PLpro) are the mediators of pp1a and pp1ab cleavage, which makes them essential for viral replication and attractive antiviral targets. Although there are no Mpro and PLpro homologues in the EBOV genome, proteases are key molecules for other viruses such as the hepatitis C virus (HCV) and the human immunodeficiency virus type 1 (HIV-1)[43]. That was the reason why it was initially thought that repurposing of HCV and HIV-1 protease inhibitors could help to treat SARS-CoV-2 infection, but unfortunately this strategy failed to provide solid therapeutic candidates[44].
EBOV and SARS-CoV-2 protein synthesis is accompanied by the replication of the viral genome (Figure 1). In EBOV infection, the L polymerase copies the negative-sense RNA generating positive-sense antigenomes, which in turn serve as templates for the synthesis of new negative-sense genomes[36]. Similarly, the RNA-dependent RNA polymerase nsp12 generates full-length negative-sense copies of SARS-CoV-2 RNA, that can be copied for generating the new positive-sense genomes[45]. Therefore, both EBOV and SARS-CoV-2 rely on the activity of their RNA-dependent RNA polymerases as central molecules for viral replication. Thus, polymerases have been also considered major targets in the development of novel antiviral therapies for both viruses.
In addition to viral proteins, host factors play a role in the transcription/replication of viral genomes. For EBOV, the DNA topoisomerase I and the RNA-binding protein Staufen 2 participate in the synthesis of viral RNAs[46][47]. NXF1 and DDX39 are RNA splicing and export factors that contribute to viral transcription and translation[48], while the protein phosphatases 1 (PP1) and 2A (PP2A) activate VP30 through dephosphorylation[49][50]. Intriguingly, the host retinoblastoma-binding protein 6 (RBBP6) and the double stranded RNA-binding protein 76 (DRBP76) are host restriction factors that inhibit PP2A and L protein activity, respectively[47][51][52], which suggests the therapeutic potential of inhibiting these viral proteins. Although there is still a lack of information regarding host factors governing SARS-CoV-2 replication, knowledge gathered in the study of other coronavirus species could provide clues on the factors involved in SARS-CoV-2 replication. For example, some coronaviruses modify the phosphorylation of the eukaryotic initiation factor 2 (eIF2) to take control over host translation [53], highlighting the therapeutic potential of inhibiting this and other translation factors. The eukaryotic elongation factor 1A2 (eEF1A2) is also a very interesting candidate that has offered a new antiviral approach. Future studies will identify novel factors involved in SARS-CoV-2 transcription/replication, thus increasing the opportunity for therapeutic interventions.
Both EBOV and SARS-CoV-2 replicate in particular cellular localizations. EBOV replicates in inclusion bodies whose formation relies on the presence of viral NP and host importin-α7[54]. Similarly, coronavirus replication occurs in specialized compartments termed replication organelles formed in the presence of nsp3, nsp4, and nsp6 viral proteins[55][56][57]. The connection between EBOV-driven inclusion bodies and the replication organelles observed in SARS-CoV-2-infected cells remains uncertain. However, the later seem to play an important role in SARS-CoV-2 replication[58], so further research in this field is guaranteed.
Taken together, a number of viral and host factors play key roles during the process of viral binding, internalization, transcription and replication of SARS-CoV-2 (Figure 1). Intriguingly, some of them have homologous counterparts in the infection by EBOV and other viruses, such as the host cathepsins and the viral polymerase and proteases. Therefore, tackling these factors could prevent productive viral infection, leading to the identification of potential broad-spectrum antiviral strategies.
References
- Moller-Tank, S.; Maury, W. Phosphatidylserine Receptors: Enhancers of Enveloped Virus Entry and Infection. Virology 2014, 468, 565–580.
- Moller-Tank, S.; Albritton, L.M.; Rennert, P.D.; Maury, W. Characterizing Functional Domains for TIM-Mediated Enveloped Virus Entry. J. Virol. 2014, 88, 6702–6713.
- Miller, M.E.; Adhikary, S.; Kolokoltsov, A.A.; Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. J. Virol. 2012, 86, 7473–7483.
- Perez-Zsolt, D.; Erkizia, I.; Pino, M.; García-Gallo, M.; Martin, M.T.; Benet, S.; Chojnacki, J.; Fernández-Figueras, M.T.; Guerrero, D.; Urrea, V.; et al. Anti-Siglec-1 Antibodies Block Ebola Viral Uptake and Decrease Cytoplasmic Viral Entry. Nat. Microbiol. 2019, 4, 1558–1570.
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 2444–2447.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 1–10.
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.
- Sigrist, C.J.; Bridge, A.; Le Mercier, P. A Potential Role for Integrins in Host Cell Entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759.
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860.
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865.
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.Y.; Geng, J.-J.; et al. SARS-CoV-2 Invades Host Cells via a Novel Route: CD147-Spike Protein. BioRxiv 2020, 1–10.
- Thepaut, M.; Luczkowiak, J.; Vives, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N.; et al. DC/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and Can Be Inhibited by a Glycomimetic Antagonist. Biorxiv 2020.
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Zheng, T.; Yan, R.; Wu, P.; Xie, S.; Zhou, Q.; Huang, J.; et al. AXL Promotes SARS-CoV-2 Infection of Pulmonary and Bronchial Epithelial Cells. BioRxiv 2020.
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the Endolysosomal Host-SARS-CoV-2 Interface by Clinically Licensed Functional Inhibitors of Acid Sphingomyelinase (FIASMA) Including the Antidepressant Fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255.
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 180, 281–292.
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS Coronavirus Spike Protein via Sequential Proteolytic Cleavage at Two Distinct Sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876.
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic Conformational Changes of a Coronavirus Spike Glycoprotein Promote Membrane Fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162.
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-NCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742.
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94.
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212.
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a Highly Conserved Domain within the Severe Acute Respiratory Syndrome Coronavirus Spike Protein S2 Domain with Characteristics of a Viral Fusion Peptide. J. Virol. 2009, 83, 7411–7421.
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 Promote SARS-CoV-2 Infection of Human Small Intestinal Enterocytes. Sci. Immunol. 2020, 5, eabc3582.
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLoS Pathog. 2010, 6, e1001121.
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-like Mechanism and Subsequent Trafficking through Early and Late Endosomes. PLoS Pathog. 2010, 6, e1001110.
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola Virus Glycoprotein by the Proprotein Convertase Furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767.
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin Cleavage Potentiates the Ebola Virus Glycoprotein To Undergo a Subsequent Fusion-Relevant Conformational Change. J. Virol. 2012, 86, 364–372.
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Virology: Endosomal Proteolysis of the Ebola Virus Glycoprotein Is Necessary for Infection. Science 2005, 308, 1643–1646.
- Mingo, R.M.; Simmons, J.A.; Shoemaker, C.J.; Nelson, E.A.; Schornberg, K.L.; D’Souza, R.S.; Casanova, J.E.; White, J.M. Ebola Virus and Severe Acute Respiratory Syndrome Coronavirus Display Late Cell Entry Kinetics: Evidence That Transport to NPC1+ Endolysosomes Is a Rate-Defining Step. J. Virol. 2015, 89, 2931–2943.
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature 2011, 477, 340–343.
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 2011, 477, 344–350.
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-Pore Channels Control Ebola Virus Host Cell Entry and Are Drug Targets for Disease Treatment. Science 2015, 347, 995–998.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263.
- Banadyga, L.; Hoenen, T.; Ambroggio, X.; Dunham, E.; Groseth, A.; Ebihara, H. Ebola Virus VP24 Interacts with NP to Facilitate Nucleocapsid Assembly and Genome Packaging. Sci. Rep. 2017, 7, 1–14.
- Watt, A.; Moukambi, F.; Banadyga, L.; Groseth, A.; Callison, J.; Herwig, A.; Ebihara, H.; Feldmann, H.; Hoenen, T. A Novel Life Cycle Modeling System for Ebola Virus Shows a Genome Length-Dependent Role of VP24 in Virus Infectivity. J. Virol. 2014, 88, 10511–10524.
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.-D.; Becker, S. Comparison of the Transcription and Replication Strategies of Marburg Virus and Ebola Virus by Using Artificial Replication Systems. J. Virol. 1999, 73, 2333–2342.
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The Coding Capacity of SARS-CoV-2. Nature 2020, 589, 125–130.
- Sims, A.C.; Ostermann, J.; Denison, M.R. Mouse Hepatitis Virus Replicase Proteins Associate with Two Distinct Populations of Intracellular Membranes. J. Virol. 2000, 74, 5647–5654.
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Oronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 58–126.
- Philip, V.; Markus, G.; Jenna, K.; Stephanie, P.; Nadine, E.; Sophie, B.L.; Cedric, S.; Jasmine, P.; Hanspeter, S.; Véronique, G.; et al. Determination of Host Cell Proteins Constituting the Molecular Microenvironment of Coronavirus Replicase Complexes by Proximity-Labeling. eLife 2019, 8, e42037.
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966.
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; MacKens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1256.
- Sharma, A.; Gupta, S.P. Fundamentals of Viruses and Their Proteases. In Viral Proteases and Their Inhibitors; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–24. ISBN 978-0-12-809712-0.
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease Targeted COVID-19 Drug Discovery and Its Challenges: Insight into Viral Main Protease (Mpro) and Papain-like Protease (PLpro) Inhibitors. Bioorg. Med. Chem. 2020, 115860.
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782.
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 Interacts with Multiple Components of the Ebola Virus Ribonucleoprotein and Enhances Viral RNA Synthesis. MBio 2018, 9.
- Takahashi, K.; Halfmann, P.; Oyama, M.; Kozuka-Hata, H.; Noda, T.; Kawaoka, Y. DNA Topoisomerase 1 Facilitates the Transcription and Replication of the Ebola Virus Genome. J. Virol. 2013, 87, 8862–8869.
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A Genome-Wide SiRNA Screen Identifies a Druggable Host Pathway Essential for the Ebola Virus Life Cycle. Genome Med. 2018, 10, 58.
- Ammosova, T.; Pietzsch, C.A.; Saygideǧer, Y.; Ilatovsky, A.; Lin, X.; Ivanov, A.; Kumari, N.; Jerebtsova, M.; Kulkarni, A.; Petukhov, M.; et al. Protein Phosphatase 1-Targeting Small-Molecule C31 Inhibits Ebola Virus Replication. J. Infect. Dis. 2018, 218, S627–S635.
- Modrof, J.; Mühlberger, E.; Klenk, H.D.; Becker, S. Phosphorylation of VP30 Impairs Ebola Virus Transcription. J. Biol. Chem. 2002, 277, 33099–33104.
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Gabriel, I.; et al. Protein Interactio Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2019, 175, 1917–1930.
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A New Ebola Virus Nonstructural Glycoprotein Expressed through RNA Editing. J. Virol. 2011, 85, 5406–5414.
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host Factors in Coronavirus Replication. Curr. Top. Microbiol. Immunol. 2018, 419, 1–42.
- Gabriel, G.; Feldmann, F.; Reimer, R.; Thiele, S.; Fischer, M.; Hartmann, E.; Bader, M.; Ebihara, H.; Hoenen, T.; Feldmann, H. Importin-Α7 Is Involved in the Formation of Ebola Virus Inclusion Bodies but Is Not Essential for Pathogenicity in Mice. J. Infect. Dis. 2015, 212, S316–S321.
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. MBio 2013, 4, e00524-13.
- Lundin, A.; Dijkman, R.; Bergström, T.; Kann, N.; Adamiak, B.; Hannoun, C.; Kindler, E.; Jónsdóttir, H.R.; Muth, D.; Kint, J.; et al. Targeting Membrane-Bound Viral RNA Synthesis Reveals Potent Inhibition of Diverse Coronaviruses Including the Middle East Respiratory Syndrome Virus. PLoS Pathog. 2014, 10, e1004166.
- Oudshoorn, D.; Rijs, K.; Limpens, R.W.A.L.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and Cleavage of Middle East Respiratory Syndrome Coronavirus Nsp3-4 Polyprotein Induce the Formation of Double-Membrane Vesicles That Mimic Those Associated with Coronaviral RNA Replication. MBio 2017, 8, e01658-17.
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A Molecular Pore Spans the Double Membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398.

