 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giuseppe Perugino | + 4619 word(s) | 4619 | 2020-04-27 08:43:11 | | | |
| 2 | Rita Xu | -1755 word(s) | 2864 | 2020-04-29 03:33:46 | | | | |
| 3 | Rita Xu | -46 word(s) | 2818 | 2020-10-29 08:53:37 | | | | |
| 4 | Rita Xu | -46 word(s) | 2818 | 2020-10-29 08:54:15 | | |
Video Upload Options
The genome of living cells is continuously exposed to endogenous and exogenous attacks, and this is particularly amplified at high temperatures. Alkylating agents cause DNA damage, leading to mutations and cell death; for this reason, a class of enzymes known as alkylguanine-DNA-alkyltransferases (AGTs) protects the DNA from mutations caused by alkylating agents, in particular in the recognition and repair of alkylated guanines in O6-position. The peculiar irreversible self-alkylation reaction of these enzymes triggered numerous studies, especially on the human homologue, in order to identify effective inhibitors in the fight against cancer. In modern biotechnology, engineered variants of AGTs are developed to be used as protein tags for the attachment of chemical ligands. In the last decade, research on AGTs from (hyper)thermophilic sources proved useful as a model system to clarify numerous phenomena, also common for mesophilic enzymes. This review traces recent progress in this class of thermozymes, emphasizing their usefulness in basic research and their consequent advantages for in vivo and in vitro biotechnological applications.
1. The O6-Alkylguanine-DNA-Alkyltransferase from Saccharolobus solfataricus
In the last decade, the O6-alkylguanine-DNA-alkyltransferase from Saccharolobus solfataricus (SsOGT) has been characterized through detailed physiological, biochemical, and structural analysis. Due to its intrinsic stability, the SsOGT protein has proven to be an outstanding model for clarifying the relationships between function and structural characteristics. As all AGTs, its reaction mechanism is based on the recognition of the damaged nucleobase on DNA [1], followed by a one-step reaction, in which the alkyl group of the damaged guanine is irreversibly transferred to a cysteine residue in its active site. For these reasons, they are also called suicide or kamikaze proteins, showing a 1:1 stoichiometry of their reaction with the natural substrate. The disadvantage of this elegant catalysis is that, upon alkylation, the protein is self-inactivated and destabilized, triggering its recognition by cellular systems to be degraded by the proteasome [2].
Saccharolobus solfataricus (previously known as Sulfolobus solfataricus) is a microorganism first isolated and discovered in 1980 in the Solfatara volcano (Pisciarelli-Naples, Italy) [3], which thrives in volcanic hot springs at 80 °C and a pH 2.0–4.0 range. In order to protect its genome in these harsh conditions, S. solfataricus evolved several efficient protection and repair systems [3][4]. S. solfataricus is highly sensitive to the alkylating agent methyl methane sulfonate (MMS), showing a transient growth arrest when treated with MMS concentrations in the range of > 0.25 mM to 0.7 mM [3][4]. Interestingly, although the ogt RNA level increases after MMS treatment, the relative enzyme concentration decreases, suggesting its degradation in cells in response to the alkylating agent and, in general, to a cellular stress [4]. Under these treatment conditions, however, the protein level rises after few hours, and, in parallel, the growth of Saccharolobus starts again [4], indicating a role of SsOGT in efficient DNA repair by alkylation damage.
1.1. Innovative OGT Assays
Various assays to measure AGT activity are reported in the literature. The first methods were based on the use of oligonucleotides carrying radioactive (3H or 14C) O6-alkylguanine groups. Proteinase K digestion was then carried out to measure the levels of marked S-methyl-cysteine in the lysate in an automatic amino acid analyser [5]. A very similar, but simpler and faster radioactive assay was used in another procedure with a 32P-terminal labelled oligonucleotide containing a modified guanine in a methylation-sensitive restriction enzyme sequence (as Mbo I). The AGT DNA repair activity thereby allowed the restriction enzyme to cut [6]. This procedure was also used by Ciaramella’s group to identify for the first time the activity of SsOGT [4]. This test has the advantage of analysing the digested fragment directly by electrophoresis on a polyacrylamide gel [7].
It was therefore improved in terms of precision by the subsequent separation of the digested oligonucleotides by HPLC. The chromatographic separation allowed the calculation of the concentration of active AGT after measuring the radioactivity of the peak corresponding to the digested fragment [7]. Similarly, Moschel’s group developed the analysis of hMGMT reaction products based on HPLC separation in 2002. This test investigated the degree of inhibition of oligonucleotides with O6-MG or O6-BG in different positions that varied from the 3’ to the 5’ end and whether they could be used as chemotherapy agents. IC50 values were obtained by quantifying the remaining active protein after the radioactive DNA reaction [8].
Although the assay measures the protein activity, the use of radioactive materials and chromatographic separations made these assays long, tedious, and unsafe.
An alternative approach was proposed in 2010 by the group of Carme Fàbrega, who set up an assay based on the thrombin DNA aptamer (TBA), a single-stranded 15 mer DNA oligonucleotide identified via Systematic Evolution of Ligands by EXponential enrichment (SELEX), which in its quadruplex form binds thrombin protease with high specificity and affinity [9]. In this assay, they put a fluorophore and a quencher to the TBA—the quadruplex structure of this oligonucleotide is compromised if a central O6-MG is present, preventing the two probes to stay closer. An AGT’s repair activity on the oligonucleotide allows the folding of the quadruplex structure and the Förster Resonance Energy Transfer (FRET) energy transfer takes place, resulting in a decrease of the fluorescence intensity [10].
Recently, the introduction of fluorescent derivatives of the O6-BG (as SNAP Vista Green, New England Biolabs) made possible the development of a novel DNA alkyl-transferase assay. Because AGT covalently binds a benzyl-fluorescein moiety of its substrate after reaction, it is possible to immediately load the protein product on a SDS-PAGE—the gel-imaging analysis of the fluorescence intensity gives a direct measure of the protein activity because of the 1:1 stoichiometry of protein/substrate (Figure 1). Signals of fluorescent protein (corrected by the amount of loaded protein by Coomassie staining analysis) obtained at different times are plotted, and a second order reaction rate is determined [4][11][12][13][14][15]. This method can be applied to all AGTs that bind O6-BG, with the exception of the E. coli Ada-C [16][17].
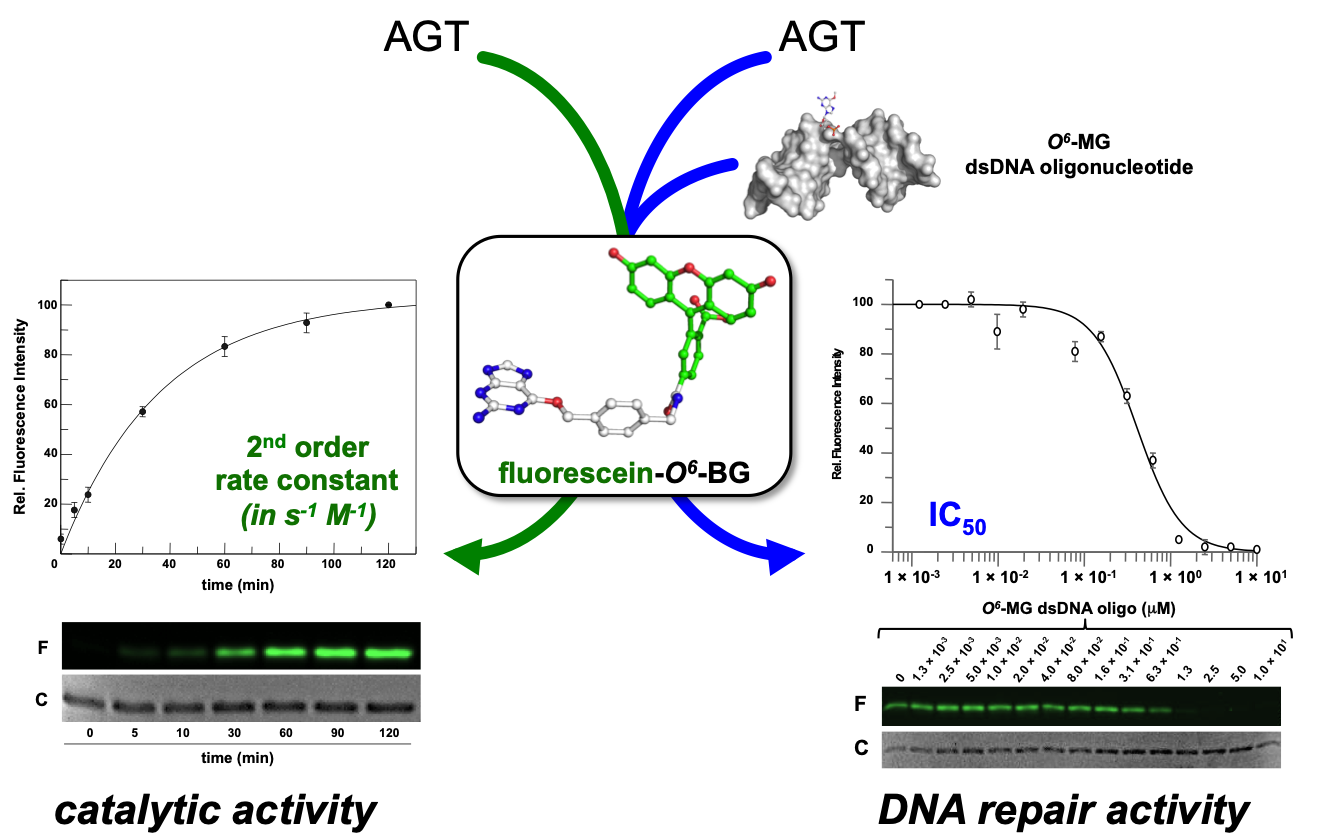
Figure 1. Innovative fluorescent AGT assay. The substrate could be used alone for the determination of the AGT catalytic activity, or in combination with a competitive non-fluorescent substrate (alkylated-DNA). In the latter case, an indirect measure of the DNA repair activity on natural substrates is determined (adapted from [18]).
Furthermore, an alkylated double strand DNA (dsDNA) oligonucleotide can be included in a competition assay with the fluorescein substrate. This non-fluorescent substrate lowers the final fluorescent signal on gel imaging analysis, depending on its concentration. In this way, it is possible to measure the activity of AGTs for their natural substrate, giving an indirect measure of methylation repair efficiency (Figure 3) [4][11][12][13][14][15]. By using this methodology, it was even possible to discriminate the SsOGT activity regarding the position of the O6-MG on DNA (see below; [12]), in line with previous data on hMGMT [19].
1.2. Biochemical Properties of S. solfataricus OGT
The recombinant SsOGT protein, heterologously expressed in E. coli, has been fully characterized using the fluorescent assay described and some results are compiled in Table 1. In agreement with its origin, the protein showed optimal catalytic activity at 80 °C, although retaining a residual activity at lower temperatures (Table 1), and in a pH range between 5.0 and 8.0. As for the most part of many thermophilic enzymes, SsOGT is resistant over a wide range of reaction conditions, such as ionic strength, organic solvents, common denaturing agents, and proteases [4][14]. Interestingly, chelating agents do not affect the activity of this enzyme. Crystallographic data clarified this observation, as the archaeal enzyme lacks a zinc ion in the structure [12], whereas this ion is important for correct folding of hMGMT [20].
Table 1. Biochemical properties comparison among SNAP-tag, SsOGT and the relative H5 mutant.
| Features | SNAP-tag® | SsOGT | SsOGT-H5 | |
|---|---|---|---|---|
| Molecular weight (kDa) a | 23.0 | 17.0 | 17.0 | |
| Topt (°C) | 37.0 | 80.0 | 75.0 | |
| Relative activity | at 25.0°C | 80% | 25% | 50%b |
| at 37.0°C | 100% | 45% | 65% | |
| at 80°C | - | 100% | 95% | |
| Catalytic activity at 37 °C (M−1 s−1) | 2.8 × 104 | 2.8 × 103 | 1.6 × 104 | |
| pHopt | 6.0 | 7.5 | 6.0 | |
| Thermal stability T½ (°C) | 6 h (37) | 3 h (70) | 3 h (70) | |
| Thermal stability T½ at 37 °C (h) | 6 | >24 | >24 | |
| Additives | NaCl | <0.3 M | >1.0 M | >1.0 M |
| EDTA | no | yes | yes | |
| sarcosyl | no | >0.5% | >0.5% | |
| DDT | yes | no | no | |
1.3. Crystal Structure of SsOGT
All catalytic steps of the AGTs’ activity (alkylated DNA recognition, DNA repair, irreversible trans-alkylation of the catalytic cysteine, recognition, and degradation of the alkylated protein) have been structurally characterized. Most information comes from the classic studies on hMGMT, as well as the Ada-C and OGT from Escherichia coli [1][20][21][22][23]. Other AGTs’ structures are also available in the Protein Data Bank site (http://www.rcsb.org/pdb/results/results.do?tabtoshow=Current&qrid=D3B02F3B).
All AGTs are inactivated after the reaction and degraded via proteasome, whereas in higher organisms, the degradation is preceded by protein ubiquitination [2]. It is a common view that the recognition of alkylated-AGTs is due by a conformational change; however, data on structure and properties of alkylated AGTs are limited because alkylation greatly destabilizes their folding [12]. The methylated-hMGMT and benzylated-hMGMT 3D structures were only obtained by flash-frozen crystals, showing that alkylation of the catalytic cysteine (C145) induces subtle conformational changes [20][24]. Consequently, these structures might not reflect the physiological conformation of the alkylated hMGMT [12].
Concerning the interactions with the DNA, SsOGT binds methylated oligonucleotides. However, the repair activity depends on the position of the alkyl-group [12]. To efficiently repair the alkylated base on DNA double helix, the protein requires at least three bases from either the 5′ or the 3′ end. This is due to the necessary interactions formed with the double helix. Structural analysis confirmed these data [12].
To overcome the serious limitation to obtain structural data from mesophilic AGTs after reaction, studies have moved to thermostable homologues, based also on the knowledge that all AGTs share a common CTD domain structure. In contrast to the human counterpart, alkylated SsOGT was soluble and relatively stable, thus allowing in-deep analysis of the protein in its post-reaction form [12]. Structural and biochemical analysis of the archaeal OGT, as well as after the reaction with a bulkier adduct in the active site (benzyl-fluorescein; [25]), suggested a possible mechanism of alkylation-induced SsOGT unfolding and degradation (Figure 2).
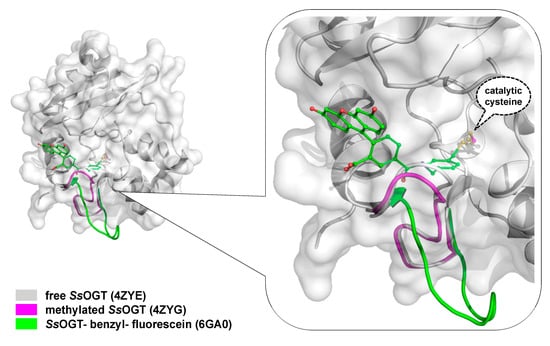
On the basis of their data, Perugino and co-workers suggested a general model for the mechanism of post-reaction AGT destabilization—the so called active-site loop moves towards the bulk solvent as a result of the covalent binding of alkyl adduct on the catalytic cysteine and the extent of the loop movement and dynamic correlates with the steric hindrance of the adduct [12][25] (Figure 2). The destabilization of this protein region triggers then the recognition of the alkylated protein by degradation pathway.
1.4. Biotechnological Applications of an Engineered SsOGT: the H5 Mutant
The introduction of the SNAP-tag technology enabled a wide in vivo and in vitro labelling variety for biological studies by fusing any protein of interest (POI) to this protein tag [26]. However, being originated from hMGMT, the extension to extremophilic organisms and/or harsh reaction conditions is seriously limited.
By following the same approach used for the hMGMT as Kai Johnsson [27][28][29][30][31], an engineered version of SsOGT was produced [4][14]. This protein, called SsOGT-H5, contains five mutations in the helix-turn-helix domain, abolishing any DNA-binding activity [4]. In addition, a sixth mutation was made—in the active site loop, where serine residue was replaced by a glutamic acid at position 132 (S132E). This modification increased the catalytic activity of SsOGT [4][14], as it was observed in the engineered version of the hMGMT during the SNAP-tag development [27]. SsOGT-H5 shows slightly lower heat stability in respect to the wild-type protein (Table 3), whereas the resistance to other denaturing agents is maintained. Moreover, SsOGT-H5 is characterised by a surprisingly high catalytic activity at lower temperatures, keeping the rate of reaction to the physiological ones (Table 3) [4][14]. These characteristics make this mutant a potential alternative to SNAP-tag for in vivo and in vitro biotechnological applications. The stability against thermal denaturation allowed Miggiano and co-workers to obtain the structure of the protein after the reaction with the fluorescent substrate SNAP-Vista Green, revealing the peculiar destabilization of the active site loop after the alkylation of the active cysteine [25].
1.4.1. In vitro Thermostable H5-Based Chimeras
The Saccharolobus OGT mutant has been firstly tested as protein tag fused to two thermostable S. solfataricus proteins heterologously expressed in E. coli. The chimeric proteins were correctly folded, and the tag did not interfere with the enzymatic activity of the tetrameric S. solfataricus β-glycosidase (Ssβgly) [14], nor with the hyperthermophile-specific DNA topoisomerase reverse gyrase [32][33][34][35][36]. Furthermore, the stability of H5 made possible a heat treatment of the cell-free extract to remove most of the E. coli proteins and performing the β-glycosidase assay at high temperatures without the need of removing the tag [15].
1.4.2. Expression in Thermophilic Organisms Models
As the applicability of the thermostable tag under in vivo conditions is very important, the SsOGT-H5 was also expressed in thermophilic organisms. The fluorescent AGT assay allows for the detection of the presence of SsOGT-H5 both in living cells as well as in vitro in cell-free extracts [14][36]. To assay the activity to SsOGT-H5, it was necessary to choose models in which the endogenous AGT activity is suppressed. Thermus thermophilus is an ogt- species, showing only one agt homologue (TTHA1564), whose annotation corresponds to an alkyltransferase-like protein (ATL) [37]. ATLs are a class of proteins present in prokaryotes and lower eukaryotes [38], presenting aminoacidic motifs similar to those of AGTs’ CTD, in which a tryptophan residue replaces the cysteine in the active site [39]. Like AGTs, ATLs use a helix-turn-helix motif to bind the minor groove of the DNA, but they do not repair it as they only recruit and interact with proteins involved in the nucleotide excision repair system [40][41].
Although T. thermophilus is a natural ogt knockout organism, Sulfolobus islandicus possesses an ogt gene very similar to that of S. solfataricus, which was silenced by a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based technique and then used as a host organism [36].
The fluorescent signal obtained by SDS-PAGE gel imaging revealed that SsOGT-H5 not only is efficiently expressed in these thermophilic microorganisms, but it also showed that this tag was correctly folded and active, demonstrating the fact that SsOGT-H5 might be used as an in vivo protein tag at high temperatures [14][36]. As is the case with SNAP-tag in human cells, the utilization of SsOGT-H5 with different fluorescent substrates gives the opportunity to perform a multi-colour fluorescence study, by following a POI inside living “thermo cells” at different stages and localization.
1.4.3. The ASLtag System
As most biotechnological processes require harsh operational conditions, the immobilization of very robust enzymes on solid supports is often essential [42]. By definition, an immobilized enzyme is a “physically confined biocatalyst, which retains its catalytic activity and can be used repeatedly” [43]. Protein immobilisation offers several advantages, such as the catalysts’ recovery and reuse, as well as the physical separation of the enzymes from the reaction mixture. Currently, different immobilisation strategies are available, from physical adsorption to covalent coupling [44][45][46][47]. However, all these procedures require purified biocatalysts and suffer from problems related to steric hindrance between the catalyst, the substrate, and the solid support, with increasing of costs and time for the production processes.
The introduction of “cell-based” immobilisation systems resulted in a significant improvement and reduces both time and costs of the process. One of the most widely used display strategies is the simultaneous heterologous expression of enzymes and their in vivo immobilisation on the external surface of Gram-negative bacteria cells, by the utilisation of the ice nucleation protein (INP) from Pseudomonas syringae [48][49]. Most recently, the N-terminal domain of INP (INPN) was used to produce a novel anchoring and self-labelling protein tag (hereinafter ASLtag). The ASLtag consists of two moieties, the INPN and the engineered and SsOGT-H5 mutant (Figure 3) [50].
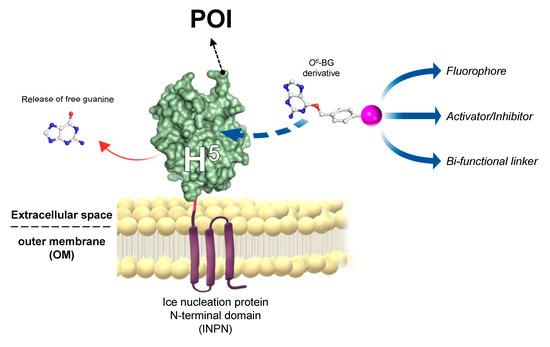
Figure 3. The novel anchoring and self-labelling protein tag (ASLtag) system. A protein of interest (POI) is genetically encoded with the tag, which in turn makes it anchored in the outer membrane and accessible for the covalent linkage to a desired chemical group (magenta sphere) by the activity of SsOGT-H5 (adapted from [51]).
The INPN allows an in vivo immobilisation on E. coli outer membrane of enzymes of interest and their exposition to the solvent. The significant reduction of the costs related to the purification and immobilization is added to the overcoming of problems related to the recovery of enzymes by simple filtration or centrifugation methods [52]. SsOGT-H5, in turn, gives the unique opportunity to label immobilized enzymes with any desired chemical groups (opportunely conjugated to the benzyl-guanine; in magenta in Figure 3) [14][28], dramatically expanding biotechnological applications of this new tool. Depending of the chemical group of choice, modulating the activity of enzymes fused with the ASLtag can be possible by introducing activator or inhibitor molecules (Figure 5). The ASLtag system was successfully employed for the expression and immobilization of monomeric biocatalysts, such as the thermostable carbonic anhydrase from Sulfurihydrogenibium yellowstonense (SspCA), as well as the tetrameric Ssβgly, without affecting their folding and catalytic activity [50]. Moreover, SspCA fused to the ASLtag showed an increase in residual activity of up to 30 % for a period of 10 days at 70 °C [51], representing a huge advantage in pushing beyond reactions in bioreactors and in the reutilization of biocatalysts.
References
- Douglas Daniels; Tammy T Woo; Kieu X Luu; David M Noll; Neil D Clarke; Anthony E Pegg; John A. Tainer; DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nature Structural & Molecular Biology 2004, 11, 714-720, 10.1038/nsmb791.
- Meng Xu-Welliver; Anthony E Pegg; Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase.. Carcinogenesis 2002, 23, 823-830, 10.1093/carcin/23.5.823.
- Anna Valenti; Alessandra Napoli; Maria Carmina Ferrara; Marc Nadal; Mosè Rossi; Maria Ciaramella; Selective degradation of reverse gyrase and DNA fragmentation induced by alkylating agent in the archaeon Sulfolobus solfataricus. Nucleic Acids Research 2006, 34, 2098-2108, 10.1093/nar/gkl115.
- Perugino, G.; Vettone, A.; Illiano, G.; Valenti, A.; Ferrara, M.C.; Rossi, M.; Ciaramella, M; Activity and regulation of archaeal DNA alkyltransferase: Conserved protein involved in repair of DNA alkylation damage. J. Biol. Chem. 2012, 287, 4222–4231, 10.1074/jbc.A111.308320.
- Olsson, M.; Lindahl, T; Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J. Biol. Chem. 1980, 255, 10569–10571.
- Wu, R.S.; Hurst-Calderone, S.; Kohn, K.W; Measurement of O6-alkylguanine-DNA alkyltransferase activity in human cells and tumor tissues by restriction endonuclease inhibition. Cancer Research 1987, 47, 6229–6235.
- Klein, S.; Oesch, F; Measurement of O6-alkylguanine-DNA alkyltransferase activity in human cells and tumor tissues by restriction endonuclease inhibition.Assay for O6-alkylguanine-DNA-alkyltransferase using oligonucleotides containing O6-methylguanine in a BamHI recognition site as substrate. Anal. Biochem. 1992, 205, 294–299.
- Luu, K.X.; Kanugula, S.; Pegg, A.E.; Pauly, G.T.; Moschel, R.C. Repair of oligodeoxyribonucleotides by O(6)-alkylguanine-DNA alkyltransferase. Biochemistry 2002, 41, 8689–8697.
- Louis C. Bock; Linda C. Griffin; John A. Latham; Eric H. Vermaas; John J. Toole; Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564-566, 10.1038/355564a0.
- Maria Tintoré; Ana Aviñó; Federico M. Ruiz; Ramón Eritja; Carme Fabrega; Development of a Novel Fluorescence Assay Based on the Use of the Thrombin-Binding Aptamer for the Detection of O6-Alkylguanine-DNA Alkyltransferase Activity. Journal of Nucleic Acids 2010, 2010, 632041, 10.4061/2010/632041.
- Riccardo Miggiano; Valentina Casazza; Silvia Garavaglia; Maria Ciaramella; Giuseppe Perugino; Menico Rizzi; Franca Rossi; Biochemical and Structural Studies of the Mycobacterium tuberculosis O6-Methylguanine Methyltransferase and Mutated Variants. Journal of Bacteriology 2013, 195, 2728-2736, 10.1128/JB.02298-12.
- Giuseppe Perugino; Riccardo Miggiano; Mario Serpe; Antonella Vettone; Anna Valenti; Samarpita Lahiri; Franca Rossi; Mosè Rossi; Menico Rizzi; Maria Ciaramella; et al. Structure-function relationships governing activity and stability of a DNA alkylation damage repair thermostable protein. Nucleic Acids Research 2015, 43, 8801-16, 10.1093/nar/gkv774.
- Riccardo Miggiano; Giuseppe Perugino; Maria Ciaramella; Mario Serpe; Dominik Rejman; Ondřej Páv; Radek Pohl; Silvia Garavaglia; Samarpita Lahiri; Menico Rizzi; Franca Rossi; Crystal structure of Mycobacterium tuberculosis O6-methylguanine-DNA methyltransferase protein clusters assembled on to damaged DNA. Biochemical Journal 2016, 473, 123-133, 10.1042/bj20150833.
- Antonella Vettone; Mario Serpe; Aurelio Hidalgo; José Berenguer; Giovanni Del Monaco; Anna Valenti; Mosè Rossi; Maria Ciaramella; Giuseppe Perugino; A novel thermostable protein-tag: optimization of the Sulfolobus solfataricus DNA- alkyl-transferase by protein engineering. Extremophiles 2016, 20, 1-13, 10.1007/s00792-015-0791-9.
- Castrese Morrone; Riccardo Miggiano; Mario Serpe; Alberto Massarotti; Anna Valenti; Giovanni Del Monaco; Mosè Rossi; Franca Rossi; Menico Rizzi; Giuseppe Perugino; Maria Ciaramella; Interdomain interactions rearrangements control the reaction steps of a thermostable DNA alkyltransferase. Biochimica et Biophysica Acta (BBA) - General Subjects 2017, 1861, 86-96, 10.1016/j.bbagen.2016.10.020.
- R H Elder; G P Margison; J A Rafferty; Differential inactivation of mammalian and Escherichia coli O6-alkylguanine-DNA alkyltransferases by O6-benzylguanine.. Biochemical Journal 1994, 298, 231-235.
- Karina Goodtzova; Sreenivas Kanugula; Suvarchala Edara; Gary T. Pauly; Robert C. Moschel; Anthony E. Pegg; Repair ofO6-Benzylguanine by theEscherichia coliAda and Ogt and the HumanO6-Alkylguanine-DNA Alkyltransferases. Journal of Biological Chemistry 1997, 272, 8332-8339, 10.1074/jbc.272.13.8332.
- Riccardo Miggiano; Anna Valenti; Franca Rossi; Menico Rizzi; Giuseppe Perugino; Maria Ciaramella; Every OGT Is Illuminated … by Fluorescent and Synchrotron Lights. International Journal of Molecular Sciences 2017, 18, 2613, 10.3390/ijms18122613.
- Melikishvili, M.; Rasimas, J.J.; Pegg, A.E.; Fried, M.G. Interactions of human O(6)-alkylguanine-DNA alkyltransferase (AGT) with short double-stranded DNAs. Biochemistry 2008, 47, 13754–13763.
- Douglas Daniels; Clifford D. Mol; Andrew S. Arvai; Sreenivas Kanugula; Anthony E. Pegg; John A. Tainer; Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. The EMBO Journal 2000, 19, 1719-1730, 10.1093/emboj/19.7.1719.
- Erica M. Duguid; Phoebe A. Rice; Chuan He; The Structure of the Human AGT Protein Bound to DNA and its Implications for Damage Detection. Journal of Molecular Biology 2005, 350, 657-666, 10.1016/j.jmb.2005.05.028.
- Julie L. Tubbs; Anthony E. Pegg; John A. Tainer; DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair 2007, 6, 1100-1115, 10.1016/j.dnarep.2007.03.011.
- M H Moore; J M Gulbis; E J Dodson; B Demple; P C Moody; Crystal structure of a suicidal DNA repair protein: the Ada O6-methylguanine-DNA methyltransferase from E. coli. The EMBO Journal 1994, 13, 1495-1501.
- Elizabeth Brunk; Birgit Mollwitz; Ursula Rothlisberger; Mechanism to Trigger Unfolding in O6-Alkylguanine-DNA Alkyltransferase. ChemBioChem 2013, 14, 703-710, 10.1002/cbic.201200566.
- Franca Rossi; Castrese Morrone; Alberto Massarotti; Davide M. Ferraris; Anna Valenti; Giuseppe Perugino; Riccardo Miggiano; Crystal structure of a thermophilic O6-alkylguanine-DNA alkyltransferase-derived self-labeling protein-tag in covalent complex with a fluorescent probe. Biochemical and Biophysical Research Communications 2018, 500, 698-703, 10.1016/j.bbrc.2018.04.139.
- Antje Keppler; Horst Pick; Claudio Arrivoli; Horst Vogel; Kai Johnsson; Labeling of fusion proteins with synthetic fluorophores in live cells. Proceedings of the National Academy of Sciences 2004, 101, 9955-9959, 10.1073/pnas.0401923101.
- Alexandre Juillerat; Thomas Gronemeyer; Antje Keppler; Susanne Gendreizig; Horst Pick; Horst Vogel; Kai Johnsson; Directed Evolution of O6-Alkylguanine-DNA Alkyltransferase for Efficient Labeling of Fusion Proteins with Small Molecules In Vivo. Chemistry & Biology 2003, 10, 313-317, 10.1016/s1074-5521(03)00068-1.
- Antje Keppler; Susanne Gendreizig; Thomas Gronemeyer; Horst Pick; Horst Vogel; Kai Johnsson; A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nature Biotechnology 2003, 21, 86-89, 10.1038/nbt765.
- Maik Kindermann; Nathalie George; Nils Johnsson; Kai Johnsson; Covalent and Selective Immobilization of Fusion Proteins. Journal of the American Chemical Society 2003, 125, 7810-7811, 10.1021/ja034145s.
- Thomas Gronemeyer; Christopher Chidley; Alexandre Juillerat; Christian Heinis; Kai Johnsson; Directed evolution of O6-alkylguanine-DNA alkyltransferase for applications in protein labeling. Protein Engineering, Design, and Selection 2006, 19, 309-316, 10.1093/protein/gzl014.
- Marlon J. Hinner; Kai Johnsson; How to obtain labeled proteins and what to do with them. Current Opinion in Biotechnology 2010, 21, 766-776, 10.1016/j.copbio.2010.09.011.
- Anna Valenti; Giuseppe Perugino; Anna D’Amaro; Andrea Cacace; Alessandra Napoli; Mosè Rossi; Maria Ciaramella; Dissection of reverse gyrase activities: insight into the evolution of a thermostable molecular machine.. Nucleic Acids Research 2008, 36, 4587-97, 10.1093/nar/gkn418.
- Anna Valenti; Giuseppe Perugino; Takehiko Nohmi; Mosè Rossi; Maria Ciaramella; Inhibition of translesion DNA polymerase by archaeal reverse gyrase.. Nucleic Acids Research 2009, 37, 4287-95, 10.1093/nar/gkp386.
- Giuseppe Perugino; Anna Valenti; Anna D'amaro; Mosè Rossi; Maria Ciaramella; Reverse gyrase and genome stability in hyperthermophilic organisms. Biochemical Society Transactions 2009, 37, 69-73, 10.1042/bst0370069.
- Anna Valenti; Giuseppe Perugino; Mosè Rossi; Maria Ciaramella; Positive supercoiling in thermophiles and mesophiles: of the good and evil. Biochemical Society Transactions 2011, 39, 58-63, 10.1042/bst0390058.
- Valeria Visone; Wenyuan Han; Giuseppe Perugino; Giovanni Del Monaco; Qunxin She; Mosè Rossi; Anna Valenti; Maria Ciaramella; In vivo and in vitro protein imaging in thermophilic archaea by exploiting a novel protein tag. PLOS ONE 2017, 10, e0185791, 10.1371/journal.pone.0185791.
- Rihito Morita; Noriko Nakagawa; Seiki Kuramitsu; Ryoji Masui; An O6-methylguanine-DNA Methyltransferase-like Protein from Thermus thermophilus Interacts with a Nucleotide Excision Repair Protein. The Journal of Biochemistry 2008, 144, 267-277, 10.1093/jb/mvn065.
- Orlando D. Schärer; Alkyltransferase-like proteins: brokers dealing with alkylated DNA bases. Molecular Cell 2012, 47, 3-4, 10.1016/j.molcel.2012.06.030.
- Tubbs, J.L.; Latypov, V.; Kanugula, S.; Butt, A.; Melikishvili, M.; Kraehenbuehl, R.; Fleck, O.; Marriott, A.; Watson, A.J.; Verbeek, B; et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature 2009, 459, 808-813, 10.1038/nature08076.
- Latypov, V.F.; Tubbs, J.L.; Watson, A.J.; Marriott, A.S.; McGown, G.; Thorncroft, M.; Wilkinson, O.J.; Senthong, P.; Butt, A.; Arvai, A.S.; et al. Atl1 regulates choice between global genome and transcription-coupled repair of O(6)-alkylguanines. Mol Cell. 2012, 47, 50–60.
- Samarpita Lahiri; Menico Rizzi; Franca Rossi; Riccardo Miggiano; Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins: Structure, Function, and Bioinformatics 2018, 86, 98-109, 10.1002/prot.25412.
- Zhou Zhou; Martín Hartmann; Progress in enzyme immobilization in ordered mesoporous materials and related applications. Chemical Society Reviews 2013, 42, 3894, 10.1039/c3cs60059a.
- Tosa, T.; Mori, T.; Fuse, N.; Chibata, I; Studies on continuous enzyme reactions. I. Screening of carriers for preparation of water-insoluble aminoacylase. Enzymologia 1969, 31, 214–224.
- Nur Royhaila Mohamad; Nur Haziqah Che Marzuki; Nor Aziah Buang; Fahrul Huyop; Roswanira Abdul Wahab; An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnology & Biotechnological Equipment 2015, 29, 205-220, 10.1080/13102818.2015.1008192.
- Hoang Hiep Nguyen; Moonil Kim; An Overview of Techniques in Enzyme Immobilization. Applied Science and Convergence Technology 2017, 26, 157-163, 10.5757/asct.2017.26.6.157.
- Jakub Zdarta; Anne S. Meyer; Teofil Jesionowski; Manuel Pinelo; A General Overview of Support Materials for Enzyme Immobilization: Characteristics, Properties, Practical Utility. Catalysts 2018, 8, 92, 10.3390/catal8020092.
- Sirisha, V.L.; Jain, A.; Jain, A. Chapter Nine—Enzyme Immobilization: An Overview on Methods, Support Material, and Applications of Immobilized Enzymes. Adv. Food Nut. Res. 2016, 79, 179–211.
- D Gurian-Sherman; S E Lindow; Bacterial ice nucleation: significance and molecular basis.. The FASEB Journal 1993, 7, 1338–1343.
- Nelly Cochet; P. Widehem; Ice crystallization by Pseudomonas syringae.. Applied Microbiology and Biotechnology 2000, 54, 153-161, 10.1007/s002530000377.
- Rosa Merlo; Sonia Del Prete; Anna Valenti; Rosanna Mattossovich; Vincenzo Carginale; Claudiu T. Supuran; Clemente Capasso; Giuseppe Perugino; An AGT-based protein-tag system for the labelling and surface immobilization of enzymes on E. coli outer membrane. Journal of Enzyme Inhibition and Medicinal Chemistry 2019, 34, 490-499, 10.1080/14756366.2018.1559161.
- Del Prete, S.; Merlo, R.; Valenti, A.; Mattossovich, R.; Rossi, M.; Carginale, M.; Supuran, C.T.; Perugino, G.; Capasso, C. Thermostability enhancement of the α-carbonic anhydrase from Sulfurihydrogenibium yellowstonense by using the anchoring-and-selflabelling-protein-tag system (ASLtag). J. Enz. Inhib. Med. Chem. 2019, 34, 946–954.
- Sonia Del Prete; Rosa Perfetto; Mosè Rossi; Fatmah A. S. Alasmary; Sameh M. Osman; Zeid Alothman; Claudiu T. Supuran; Clemente Capasso; A one-step procedure for immobilising the thermostable carbonic anhydrase (SspCA) on the surface membrane of Escherichia coli. Journal of Enzyme Inhibition and Medicinal Chemistry 2017, 32, 1120-1128, 10.1080/14756366.2017.1355794.

