 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jing-hsiung Ou | + 3554 word(s) | 3554 | 2021-02-01 07:22:14 | | | |
| 2 | Dean Liu | -1352 word(s) | 2202 | 2021-02-02 04:37:39 | | |
Video Upload Options
Hepatitis C virus induces autophagy and temporally regulates the autophagic flux to support its replication. It uses autophagic membranes as a platform for its RNA replication, autophagic vacuoles to support its morphogenesis and autophagy to suppress host innate immune responses.
1. Introduction
Many reports have indicated that HCV can induce autophagy (i.e., macroautophagy) to enhance its own replication [1][2]. Autophagy is a catabolic process that is important for maintaining cellular homeostasis. It removes protein aggregates and damaged organelles from cells. Autophagy can be initiated by many stimuli including nutrient starvation, oxidative stress, ER stress, and microbial infections[3]. This process begins with the formation of membrane crescents, called phagophores or isolation membranes, in the cytoplasm. The membrane of phagophores will subsequently extend to form enclosed double-membrane vesicles, known as autophagosomes. Autophagosomes mature by fusing with lysosomes to form autolysosomes, in which the cargos of autophagosomes are degraded by lysosomal enzymes for recycling[4]. Alternatively, autophagosomes may also fuse with multivesicular bodies (i.e., late endosomes) to form amphisomes to positively or negatively regulate the release of exosomes from cells[5]. More than 30 autophagy-related proteins (ATGs) and two ubiquitin-like conjugation systems that are important for autophagy have been identified. Autophagy is initiated after the activation of the class III phosphatidylinositol-3-kinase (PI3KC3), which catalyzes the formation of phosphatidylinositol-3-phosphate (PI3P) to trigger the formation of the pre-autophagosomal structure (also known as the phagophore assembly site) (PAS)[6]. One ubiquitin-like conjugation system is involved in the covalent linking of ATG5 and ATG12, which will recruit ATG16 to form a complex. This ATG5–ATG12–ATG16 complex is important for the formation of phagophores. A second ubiquitin-like conjugation system is involved in the coupling of the microtubule-associated protein light-chain 3 (LC3) to phosphatidylethanolamine (PE), a phospholipid. This lipidation allows LC3 to localize to autophagosomal membranes and is important for the formation of autophagosomes. LC3 is de-lipidated by ATG4 after the maturation of autophagosomes and released back into the cytosol. It can also be degraded by lysosomal enzymes if it is localized to the inner membrane of autophagosomes[4]. Autophagy can function as a cell defense mechanism by eliminating intracellular microbial pathogens in a process known as xenophagy[7][8]. However, some microbial pathogens including viruses have developed mechanisms to subvert autophagy and use it to support their own replications. HCV has been shown to induce autophagy to enhance its own replication[1][9][10][11].
2. Mechanism of HCV-Induced Autophagy
HCV infection induces autophagy in its host cells, including hepatoma cells, primary human hepatocytes, and hepatocytes of infected individuals[9][10][12]. This is evidenced by the increase of lipidation of LC3 and the accumulation of autophagic vacuoles in HCV-infected cells. HCV induces autophagy via multiple pathways[1]. It can induce autophagy indirectly and directly. HCV has been shown to induce the ER stress and activate the unfolded protein response (UPR)[10][13][14][15][16]. The accumulation of unfolded or misfolded proteins in the ER will induce the ER stress, leading to the activation of the activating transcription factor 6 (ATF6), the inositol-requiring enzyme 1 (IRE1), and the double-stranded RNA-activated protein kinase-like ER kinase (PERK) to trigger downstream signaling pathways, which are collectively known as the UPR. The UPR is important for HCV-induced autophagy, as the silencing of ATF6, IRE1, or PERK significantly inhibits HCV-induced autophagy[10][13][17]. Further analysis indicated that the ER stress induced by HCV could inhibit AKT, also known as protein kinase B (PKB), and a negative regulator of tuberous sclerosis complex (TSC), to result in the inhibition of the mammalian target of rapamycin complex I (mTORC1). This led to the activation of the UNC-51-like kinase 1 (ULK1) and the induction of autophagy [14]. A separate study of the same group also indicated that the HCV core protein by itself was sufficient to induce the ER stress, although it activated only PERK and ATF6 without activating IRE1[17]. The activation of PERK led to the induction of the ATF4 transcription factor and the DNA damage-inducible transcript 3 protein (DDIT3, also known as CHOP). ATF4 upregulates the expression of ATG12 whereas CHOP binds to nucleotides −293 to −99 of the LC3B promoter to stimulate the expression of LC3B to induce autophagy[17]. It should be noted that in a separate study conducted by Mohl et al.[18], it was found that the lipidation of LC3 preceded detectable UPR responses and that the IRE1 knockdown did not affect the lipidation of LC3 in HCV-infected cells. Thus, Mohl et al. questioned the role of the ER stress in HCV-induced autophagy. The reason for this discrepancy of results is not clear. However, in the study by Mohl et al., the LC3 lipidation was detected at as early as 4 h post-HCV infection[18], which was in sharp contrast to the studies conducted by Huang et al., who used the same HCV JFH1 strain for the infection studies and did not detect a significant increase of LC3 lipidation until 4 days after infection[14]. In the studies of Huang et al., the authors used UV-inactivated HCV as the negative control in their infection studies to rule out the possible nonspecific effect of the inoculum. However, such control was not included in the studies of Mohl et al. and hence the possible induction of autophagy by the nonspecific effect of the inoculum could not be ruled out. It is also possible that this early increase of lipidated LC3 detected by Mohl et al. was due to the induction of Rubicon, which suppresses the maturation of autophagosomes and hence increases the level of lipidated LC3 (see below).
HCV has also been known to induce autophagy via the induction of oxidative stress[19]. HCV can induce oxidative stress through multiple mechanisms including the induction of chronic liver inflammation, iron overload, and liver injury[20]. The activities of many of its gene products that include core, E1, E2, NS4B, and NS5A proteins can also induce oxidative stress[21]. High levels of reactive oxygen species (ROS) induced by HCV cause the phosphorylation of serine-349 of the p62 sequestosome protein to induce autophagy[19]. p62 interacts with LC3 and is important for the delivery of proteins to autophagosomes for their eventual degradation in autolysosomes. The phosphorylation of p62 at serine-349 increases the affinity of p62 to Keap1, thereby disrupting the interaction between Keap1 and the nuclear factor E2-related factor 2 (Nrf2). This leads to the nuclear localization of Nrf2 and the activation of its target genes including many antioxidant genes. However, whether HCV can activate Nrf2 is controversial. While one report indicated that it could[22], others have indicated that Nrf2 was retained in the cytoplasm in association with the HCV RNA replication complex and failed to localize to the nucleus to activate its target genes[19][23]. How ROS induced by HCV mediates the serine-349 phosphorylation of p62 is unclear. A previous study indicated that hVps34, the catalytic subunit of PI3KC3, could enhance the interaction between protein kinase C-δ (PKC-δ) and p62 for the phosphorylation of p62 at serine-349 in breast cancer cells[24]. However, whether this is also the case in HCV-infected cells remains to be determined, as previous studies indicated that hVps34 was dispensable for HCV-induced autophagy[25][26].
In addition to indirectly inducing autophagy, HCV can also induce autophagy directly via the activities of its proteins. The expression of the HCV NS3-NS5B nonstructural polyprotein was sufficient to induce double-membrane vesicles that resembled autophagosomes[27]. Further expression studies of individual HCV nonstructural proteins indicated that HCV NS4B protein was sufficient to induce the UPR, the lipidation of LC3, and autophagic vacuoles[28]. The NS3/4A complex, and NS5A and NS5B individually were also able to induce autophagic vacuoles, although the effects of the latter two were much less prominent[17][28]. HCV NS3 interacts with immunity-related GTPase family M protein (IRGM)[29], which is a member of the small GTPase family and can interact with multiple autophagy-associated proteins such as ATG5 and ATG10. IRGM is critical for HCV-induced autophagy, as its depletion suppressed HCV-induced autophagy. HCV NS3 may interact with ATG5 and ATG10 via IRGM to induce autophagy. Alternatively, as IRGM mediates the dephosphorylation of serine-757 of ULK1, which is a kinase important for the initiation of autophagy, in HCV-infected cells[30], it is also possible that this effect of IRGM on ULK1 is triggered by the binding of HCV NS3 to IRGM, leading to the initiation of autophagy. Further studies will be required to test these possibilities. HCV NS4B can induce the expression of Rubicon, which suppresses the fusion between autophagosomes and lysosomes (see below), and may increase the levels of lipidated LC3 and autophagosomes through this mechanism[28]. NS4B has also been shown to interact with Beclin-1, hVps34, and Rab5 to modulate autophagy[1]. Beclin-1 is a component of the PI3KC3 complex. However, as the silencing of hVps34, the catalytic subunit of PI3KC3, and the inhibition of PI3KC3 with 3-methyladenine did not abolish autophagy induced by HCV [25], whether the interaction between NS4B, Beclin-1, and hVps34 plays any role in HCV-induced autophagy remains to be determined. In this regard, it is interesting to note that the HCV p7 ion channel protein had also been found to bind to Beclin-1 without inducing any autophagic response[31].
The molecular mechanisms of HCV-induced autophagy, including direct and indirect effects, are illustrated in Figure 1.
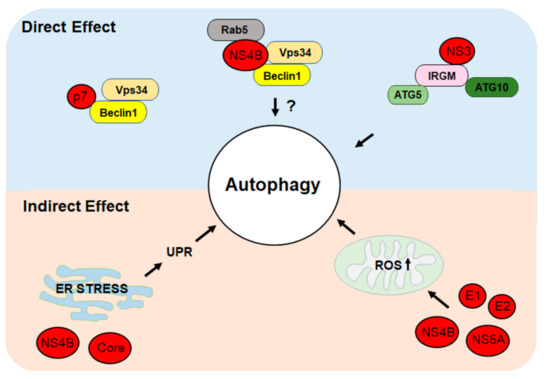
Figure 1. Direct and indirect mechanisms of hepatitis C virus (HCV)-induced autophagy. HCV proteins can directly induce autophagy by interacting with cellular proteins that regulate autophagy. HCV can also indirectly induce autophagy by inducing the ER stress to trigger the unfolded protein response (UPR) or by inducing the production of reactive oxygen species (ROS) and oxidative stress. See text for details.
3. Biogenesis of Autophagosomes Induced by HCV
The biogenesis of autophagosomes is often initiated at an ER subdomain enriched in phosphatidylinositol-3-phosphate (PI3P), a product of PI3KC3. This subdomain is called the omegasome, which serves as the PAS[32]. As mentioned above, HCV infection induces the accumulation of autophagosomes in its host cells. The induction of autophagosomes was found in cells transfected with the HCV genomic RNA or infected with HCV, and in cells harboring the replicating HCV subgenomic RNA replicon[13][25][28][33][34][35]. It had previously been reported that the double-FYVE domain-containing protein 1 (DFCP1), which binds to PI3P to initiate the biogenesis of autophagosomes, is important for HCV RNA replication[26]. As the silencing of DFCP1 also led to the reduction of LC3 lipidation, this report supports the involvement of omegasomes in the initiation of autophagosomes induced by HCV. However, as mentioned above, PI3KC3 is dispensable for HCV-induced autophagy[25][26]. Thus, DFCP1 may participate in the biogenesis of HCV-induced autophagosomes via a noncanonical pathway that does not involve PI3P. By conducting live-cell imaging, our recent studies indicated that phagophores induced by HCV originated from the ER[36]. They then underwent homotypic fusion to generate autophagosomes via a pathway dependent on the SNARE protein syntaxin 7[36] (Figure 2). Curiously, the time required for phagophores to progress to autophagosomes in HCV-infected cells was approximately 30 min, whereas that for autophagy induced by nutrient deprivation took less than 10 min[36]. The reason why the biogenesis of autophagosomes in HCV-infected cells requires a much longer period of time is unclear, possibly being due to the involvement of homotypic fusion of phagophores and/or other biological processes specific to HCV. HCV infection causes the fragmentation of Golgi membranes in an IRGM-dependent manner[30]. Interestingly, these fragmented Golgi membranes were found to colocalize with LC3 puncta (i.e., autophagosomes), the ER marker, and the replicating HCV RNA, suggesting that Golgi membranes may also be involved in the biogenesis of autophagosomes[30].
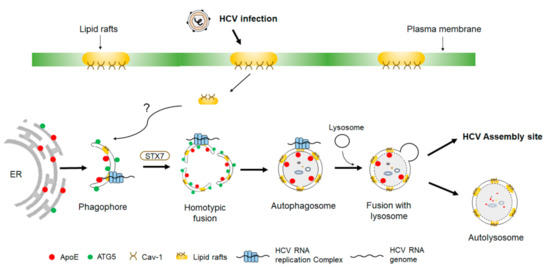
Figure 2. Biogenesis of autophagosomes induced by HCV. Phagophores induced by HCV originate from the ER membranes. The HCV RNA replication complex as well as its associated lipid rafts and caveolin-1 (Cav-1) become associated with phagophores through an unknown mechanism. Phagophores subsequently undergo homotypic fusion in a process dependent on syntaxin 7 (STX7) to form autophagosomes. During these processes, apolipoprotein E (ApoE) becomes associated with autophagosomes and is delivered by autophagosomes to the HCV assembly site to interact with the HCV E2 envelope protein. Some of the autophagosomes may also fuse with lysosomes to form autolysosomes to result in the autophagic degradation of ApoE.
Multiple studies have indicated that the fusion of autophagosomes with lysosomes is delayed in HCV-infected cells, resulting in the accumulation of autophagosomes in the early stage of HCV infection[10][25][28][37][38]. This delayed maturation of autophagosomes in HCV-infected cells was found to be due to the differential induction of Rubicon and UVRAG proteins by HCV [28][38]. Rubicon negatively regulates the maturation of autophagosomes, whereas UVRAG positively regulates it. In the early stage of HCV infection, Rubicon was upregulated, which inhibited the fusion between autophagosomes and lysosomes. However, in the late stage of infection, UVRAG was also upregulated to overcome the inhibitory effect of Rubicon, resulting in the maturation of autophagosomes and the completion of the autophagic flux[28]. Jones-Jamtgaard et al. also reported that Arl8b, an Arf-like GTPase that localizes to lysosomes and regulates the autophagic flux, was redistributed by HCV to peripheral locations to suppress the autophagosome–lysosome fusion[37]. These studies indicated that HCV could use different mechanisms to delay the maturation of autophagosomes and suppress the autophagic flux. This temporal regulation of the autophagic flux allows the accumulation of autophagosomes in the early stage of HCV infection and is beneficial to HCV replication (see below).
References
- Wang, L.; Ou, J.H. Hepatitis c virus and autophagy. Biol. Chem. 2015, 396, 1215–1222.
- Chan, S.T.; Ou, J.J. Hepatitis c virus-induced autophagy and host innate immune response. Viruses 2017, 9, 224.
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293.
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42.
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42.
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class iii pi3-kinase complexes. J. Cell Biol. 2009, 186, 773–782.
- Bauckman, K.A.; Owusu-Boaitey, N.; Mysorekar, I.U. Selective autophagy: Xenophagy. Methods 2015, 75, 120–127.
- Chandra, P.; Kumar, D. Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in mycobacterium tuberculosis-infected macrophages. Autophagy 2016, 12, 608–609.
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis c virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249.
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061.
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051.
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945.
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56.
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis c virus inhibits akt-tuberous sclerosis complex (tsc), the mechanistic target of rapamycin (mtor) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195.
- Shinohara, Y.; Imajo, K.; Yoneda, M.; Tomeno, W.; Ogawa, Y.; Kirikoshi, H.; Funakoshi, K.; Ikeda, M.; Kato, N.; Nakajima, A.; et al. Unfolded protein response pathways regulate hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 2013, 432, 326–332.
- Waris, G.; Tardif, K.D.; Siddiqui, A. Endoplasmic reticulum (er) stress: Hepatitis c virus induces an er-nucleus signal transduction pathway and activates nf-kappab and stat-3. Biochem. Pharmacol. 2002, 64, 1425–1430.
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis c virus core protein activates autophagy through eif2ak3 and atf6 upr pathway-mediated map1lc3b and atg12 expression. Autophagy 2014, 10, 766–784.
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis c virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732.
- Medvedev, R.; Ploen, D.; Spengler, C.; Elgner, F.; Ren, H.; Bunten, S.; Hildt, E. Hcv-induced oxidative stress by inhibition of nrf2 triggers autophagy and favors release of viral particles. Free Radic. Biol. Med. 2017, 110, 300–315.
- Choi, J.; Ou, J.H. Mechanisms of liver injury. Iii. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851.
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis c virus proteins activate nrf2/are pathway by distinct ros-dependent and independent mechanisms in huh7 cells. PLoS ONE 2011, 6, e24957.
- Jiang, Y.; Bao, H.; Ge, Y.; Tang, W.; Cheng, D.; Luo, K.; Gong, G.; Gong, R. Therapeutic targeting of gsk3beta enhances the nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis c. Gut 2015, 64, 168–179.
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis c virus impairs the induction of cytoprotective nrf2 target genes by delocalization of small maf proteins. J. Biol. Chem. 2011, 286, 8941–8951.
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. Vps34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862.
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus rna on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043.
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016, 97, 680–693.
- Chatterji, U.; Bobardt, M.; Tai, A.; Wood, M.; Gallay, P.A. Cyclophilin and ns5a inhibitors, but not other anti-hepatitis C virus (hcv) agents, preclude hcv-mediated formation of double-membrane-vesicle viral factories. Antimicrob. Agents Chemother. 2015, 59, 2496–2507.
- Wang, L.; Tian, Y.; Ou, J.H. Hcv induces the expression of rubicon and uvrag to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764.
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. Irgm is a common target of rna viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422.
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis c virus triggers golgi fragmentation and autophagy through the immunity-related gtpase m. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471.
- Aweya, J.J.; Mak, T.M.; Lim, S.G.; Tan, Y.J. The p7 protein of the hepatitis C virus induces cell death differently from the influenza a virus viroporin m2. Virus Res. 2013, 172, 24–34.
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244.
- Mizui, T.; Yamashina, S.; Tanida, I.; Takei, Y.; Ueno, T.; Sakamoto, N.; Ikejima, K.; Kitamura, T.; Enomoto, N.; Sakai, T.; et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 2010, 45, 195–203.
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis c virus upregulates beclin1 for induction of autophagy and activates mtor signaling. J. Virol. 2012, 86, 8705–8712.
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194.
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. Hcv-induced autophagosomes are generated via homotypic fusion of phagophores that mediate hcv rna replication. PLoS Pathog. 2017, 13, e1006609.
- Jones-Jamtgaard, K.N.; Wozniak, A.L.; Koga, H.; Ralston, R.; Weinman, S.A. Hepatitis c virus infection increases autophagosome stability by suppressing lysosomal fusion through an arl8b-dependent mechanism. J. Biol. Chem. 2019, 294, 14257–14266.
- Shiode, Y.; Hikita, H.; Tanaka, S.; Shirai, K.; Doi, A.; Sakane, S.; Kai, Y.; Nakabori, T.; Yamada, R.; Kodama, T.; et al. Hepatitis c virus enhances rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci. Rep. 2020, 10, 15290.


