 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pedro Martinez-Gomez | + 2265 word(s) | 2265 | 2020-03-11 05:06:18 | | | |
| 2 | Rita Xu | -500 word(s) | 1765 | 2020-04-28 12:20:38 | | | | |
| 3 | Pedro Martinez-Gomez | + 259 word(s) | 2024 | 2020-04-29 08:55:45 | | | | |
| 4 | Pedro Martinez-Gomez | + 295 word(s) | 2060 | 2020-04-29 09:05:53 | | | | |
| 5 | Pedro Martinez-Gomez | + 295 word(s) | 2060 | 2020-04-29 09:28:16 | | | | |
| 6 | Pedro Martinez-Gomez | + 295 word(s) | 2060 | 2020-04-29 10:00:17 | | | | |
| 7 | Rita Xu | -45 word(s) | 2015 | 2020-10-26 04:18:54 | | |
Video Upload Options
Plant breeding is a long and tedious process involving the generation of large populations through controlled crosses and the final selection of top individuals, the future new varieties. This process can take between 5 years in the case of horticultural crops to 15 years in the case of perennial fruit crops or 25 years in forest species. Plant breeding is an applied science, insofar as it is focused on solving specific problems, such as productivity, resistance to biotic and abiotic stresses, fruit quality, postharvest performance and sensorial attributes. In this context, a critical decision is the choice of genotypes that are used as parents. Additionally, the management, phenotyping and selection process of these seedlings are the main factors limiting the generation of new cultivars. In order to improve efficiency and sturdiness of plant breeding programs in relation to parent and seedling selection, the implementation of molecular tools is an essential requirement, including development of Marker Assisted Selection (MAS) strategies. On the other hand, we are facing a new molecular-biological perspective based on new methodologies that are affecting the genetics theory in addition to the definition of gene and Central Dogma of Molecular Biology (CDMB). This new molecular perspective, open new possibilities to improve the use of molecular tools in plant breeding. The goal of this review is the discussion about the new perspective of Plant Breeding in the context of the present Postgenomic era.
1. Key Challenges and Considerations in the Postgenomic era
The Post-genomic era includes features from a methodological point of view and from a global perspective. Firstly, researchers in this era have incorporated new methods of high-throughput sequencing of DNA and RNA, the development of complete genomes that allow a precise reference of the molecular results obtained, and the development of high-throughput methods as Genotype by Sequencing (GBS) for genomic analysis (SNPs, Single Nucleotide Polymorphism), and RNAseq for transcriptomic analysis (DEGs, Differentially Expressed Genes). The generation of large genetic and phenotypic information obtained in a massive way (through high-throughput sequencing and high throughput phenotyping approaches) and the creation of databases with billions of these data, result in what is called in research “Big Data”, which also require the development of new mathematical algorithms (such as machine learning models) and a large set of bioinformatic tools. In addition, from a global perspective, the center of gravity of the molecular processes is focused on the expression of genes and the way in which such expression is regulated. These features have turned researcher’s attention towards the study of gene regulatory networks, and in particular to the role of RNA and other non-derived DNA biomolecules [1].
2. Availability of reference genomes
At this moment, more than 550 plant species have sequenced and their reference genomes are available including the most important crops [2] (Table 1). This whole sequenced work started in 2002 with the development of the reference genome of the rice. Last years for instance, several important crops were sequenced including hazelnut, mulberry, pistachio, poplar or almond. The development of complete genomes is making any organism accessible and amenable for many kinds of studies [1] which will allow a precise reference of the molecular results obtained, and the development of high-throughput methods for genomic analysis involving the most abundant genetic variation (SNPs) and transcriptomic analysis at gene expression level (DEGs) in a new postgenomic perspective [3].
Table 1. Most important crops sequenced with available reference genome at https://www.ncbi.nlm.nih.gov/genome [2].
|
Species |
Crop |
Year |
Species |
Crop |
Year |
|
Kiwifruit |
2013 |
Lettuce |
2011 |
||
|
Pineapple |
2016 |
Apple |
2010 |
||
|
Peanut |
2018 |
Cassava |
2014 |
||
|
Argan |
2018 |
Mulberry |
2019 |
||
|
Asparagus |
2016 |
Tobacco |
2014 |
||
|
Oats |
2018 |
Olive |
2017 |
||
|
Sugar beet |
2013 |
Rice |
2002 |
||
|
Rape |
2014 |
Avocado |
2018 |
||
|
Cabbage |
2014 |
Bean |
2013 |
||
|
Mustard |
2011 |
Date Palm |
2009 |
||
|
Tea |
2019 |
Pine |
2015 |
||
|
Hemp |
2011 |
Pistachio |
2019 |
||
|
Pepper |
2013 |
Pea |
2018 |
||
|
Safflower |
2016 |
Poplar |
2019 |
||
|
Chickpea |
2012 |
Cherry |
2017 |
||
|
Watermelon |
2011 |
Almond |
2019 |
||
|
Clementina |
2013 |
Peach |
2011 |
||
|
Orange |
2012 |
Pomegranate |
2017 |
||
|
Coffee |
2018 |
Oak |
2018 |
||
|
Hazelnut |
2019 |
Rosa |
2018 |
||
|
Muskmelon |
2012 |
Secale cereale |
Rye |
2016 |
|
|
Cucumber |
2007 |
Tomato |
2009 |
||
|
Pumpkin |
2017 |
Eggplant |
2014 |
||
|
Carrot |
2016 |
Potato |
2011 |
||
|
Strawberry |
2010 |
Sorghum |
2009 |
||
|
Soybean |
2014 |
Cacao |
2016 |
||
|
Cotton |
2015 |
Wheat |
2010 |
||
|
Sunflower |
2017 |
Bean |
2015 |
||
|
Rubber |
2013 |
Cowpea |
2016 |
||
|
Barley |
2011 |
Grape |
2007 |
||
|
Walnut |
2015 |
Maize |
2010 |
3. New sequencing and bioinformatic methodologies
The new Big Data Biology harboring the development of DNA (and RNA as cDNA) high-throughput sequencing technologies together with bioinformatics analysis as well as the creation of databases with billions of data has made possible to access genetics knowledge at the level of each nucleotide. This new methodological perspective where millions of sequences are available in one single experiment with the detailed information of complete genomes (defined as the DNA organized into separate chromosomes inside the nucleus of a cell) and transcriptomes (described as the complete list of all types of RNA molecules). Several authors have even characterized this data-intensive biology as a new kind of science, a science of information management, different from traditional biology.
On the other hand, high-troughput sequencing technologies resulted in a great advance in the development and application of MAS strategies. The first generation of sequencers had an average cost of 0.5 euro per base or nucleotide, which was unaffordable for most laboratories. Later, second generation or high performance ("high-throughput") sequencing techniques emerged for sequencing DNA (DNA-Seq, in 2005) and cDNA from RNA (RNA-Seq, in 2008), based on generating thousands of parallel sequencing reactions immobilized on a solid surface. This technology lowered the cost of sequencing by reducing the reagent requirements. The cost of 0.01 euro per Mb in 2018 facilitated the launch of massive sequencing techniques. In addition, the development of high-throughput methods for genomic analysis (SNPs, Single Nucleotide Polymorphism) and transcriptomic analysis (DEGs, Differentially Expressed Genes) through the FLUIDIGM (https://www.fluidigm.com/) platform is another relevant progress.
This new perspective integrating available reference genomes and new sequencing and bioinformatic methodologies will allow the implementation of new MAS to accelerate breeding process and genome editing strategies [3][4].
4. New postgenomic perspectives
Additionally, from a global point of view, the new post-genomics era is characterized by a change in perspective on trait expression derived from the Encyclopedia of DNA Elements (ENCODE) project, performed on humans, in which the study of RNA rather than DNA was determined as the linchpin of trait expression processes [5]. In fact, the genomes of eukaryotic organisms are almost entirely transcribed, giving birth to an enormous number of non–protein-coding RNAs. In this new context, a recent concept, pervasive (interleaved) transcription, is being applied to the whole transcription process. These authors described pervasive transcription as the transcription of the interspersed genes that are embedded within the normal coding genes. The entire stretch of the genome (in other times called “junk DNA”) is transcribed, whether it is coding for a particular protein or not. Therefore, not all coding sequences lie juxtaposed, and they may also overlap one another. Jarvis and Robertson [6] describe this pervasive transcription of noncoding RNA as the dark matter emerging in the post-genomics era.
The new concept of the gene as a process has been applied to the ENCODE project, which has reconsidered the role of RNA compared to DNA [3]. The translation in protein of transcribed mRNA (messenger), together with the presence of non-coding RNA and non-regulatory RNA as rRNA and tRNA and post-transcriptional and post-translational regulation by small noncoding but regulating RNA (miRNA, siRNA, PiRNA or snoRNA), are key to the process of DNA transcription and expression in a particular phenotype. The new research results emphasize the importance of all types of RNA in the final expression of all DNA (including the formerly called “junk DNA”). In addition, ENCODE somewhat tipped the balance for the first time, albeit slightly, in favor of RNA, emphasizing the need for resources to go deeper into these mechanisms of life explained as processes. This new concept definitely rejects the concept of “junk DNA”. In the same vein, it is necessary to indicate the huge number of kinds of RNA with little or no coding capacity that have been discovered quite recently, suggesting that the gene transcript is not the fundamental unit operating in genetics. Moreover, Gerstein et al. [6] defined the gene as a DNA or RNA sequence encoding either directly or from overlapping regions for a functional RNA or protein.
In addition, in the post-genomics era, numerous authors have showed important experimental phenomenon affecting the following concepts: the gene (gene fusion and pleiotropy); the CDMB (copy-number variation, epigenetic regulation of DNA expression, long noncoding RNA, pervasive transcription, DNA damage); the origin of genetic variation (adaptive mutations, epigenetic inheritance or inheritance of acquired traits); and the effect of the environment on these variations (transgenerational transmission of environmental information). These phenomena are new exemplars out of the genetics paradigm. This situation has evolved at an ever-increasing speed over the last years, with significant new experimental, such as the epitranscriptomic (epigenetic regulation of RNA), the epimutations, the long noncoding RNA, the DNA damage or the transgenerational transmission of environmental information (Table 2).
Table 2. Experimental phenomena evidenced in the postgenomic era.
|
Experimental phenomenon |
Year |
Reference |
|
Adaptive mutations |
2005 |
[7] |
|
Copy-number variation |
2005 |
[8] |
|
Gene fusions |
2006 |
[9] |
|
Pleiotropy |
2006 |
[10] |
|
Epigenetic regulation of DNA expression |
2010 |
[11] |
|
Epigenetic inheritance |
2010 |
[12] |
|
Pervasive transcription |
2011 |
[13] |
|
Epigenetic regulation of RNA |
2012 |
[14] |
|
Epimutations |
2014 |
[15] |
|
Long noncoding RNA |
2015 |
[16] |
|
DNA damage |
2017 |
[17] |
|
Transgenerational transmission of environmental information |
2017 |
[18] |
5. Versatility of molecular markers application
In this context, genomic (DNA analysis including Copy-number variation, Gene fusions and Pleiotropy) studies for the development of MAS strategies are particularly useful when the evaluation of the character is expensive, time-consuming or with long juvenile periods. In addition, proteomic (proteins and enzymes), transcriptomic (mRNA, lncRNA) and epigenetic (DNA Methylation and histone modifications, epimutations) studies are being applied to breeding programs. These integrated approaches have been classically performed for the genetic characterization of the plant material. However, at this moment the development of suitable markers to be applied in selection for agronomical traits must be used for the clarification of the mentioned genomic studies and the development of more efficient markers.
These new perspectives of plant molecular breeding include the application of different developed markers (at genomic, transcriptomic and epigenetic level) in both development of new varieties and exploitation (Figure 1).
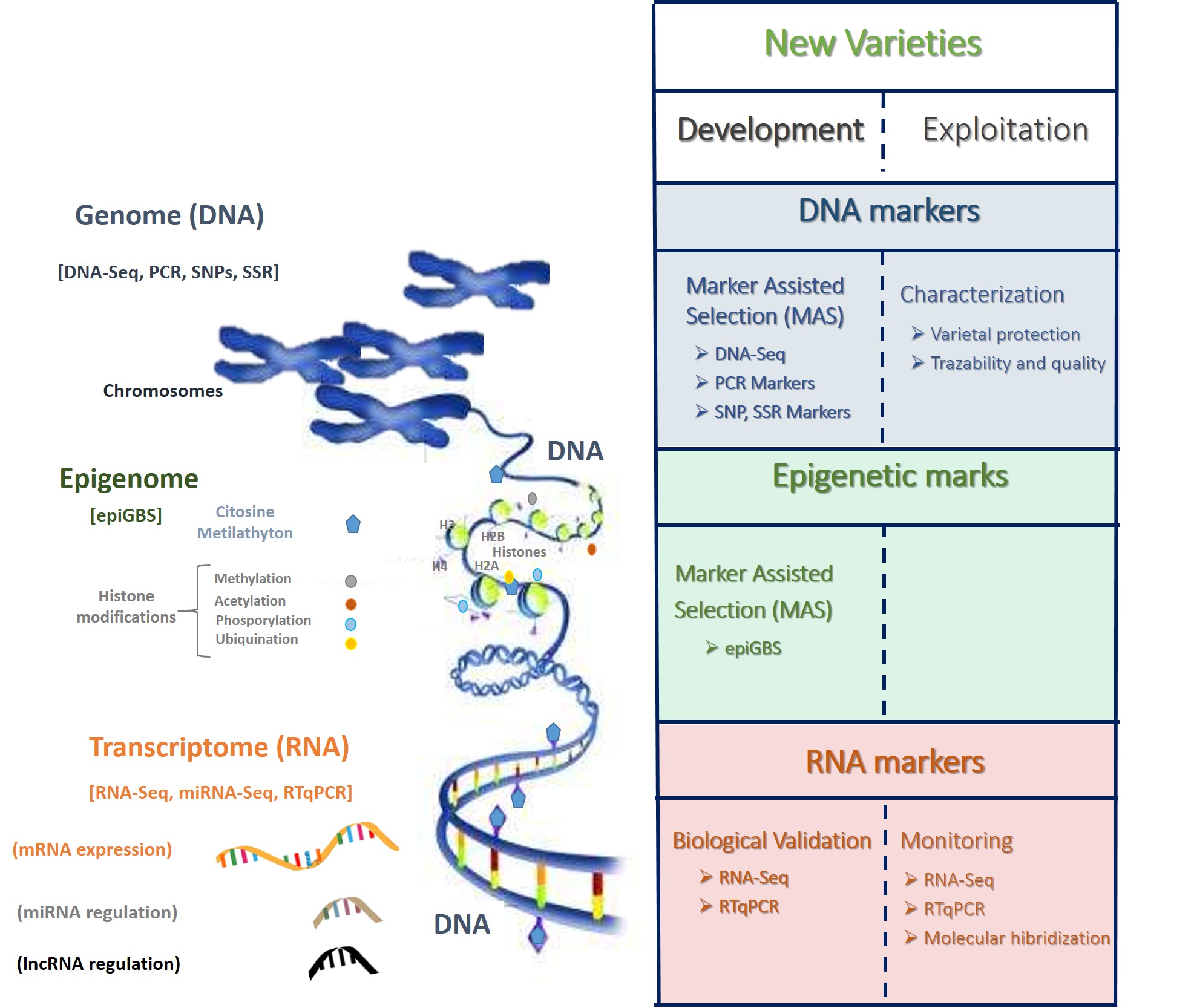
Figure 1. Schematic illustration of DNA, RNA and epigenetic markers applied in plant breeding in the development and exploitation of new varieties.
6. Future Outlook
Plant productivity for feeding an increasing human population and the study of plant diseases and their management in unpredictable context of global climatic changes are particularly relevant aspects as well as genetic plant improvement from the point of view of the quality of fruits and vegetables. Therefore, classical breeding must be combined with MAS using the diverse molecular tools available in the postgenomic era. This situation does mean that MAS will replace conventional breeding, other ways is a necessary complement. The practical learning that conventional breeding gives is unparalleled with the molecular support. In this context, there has been a significant shift of conventional breeders to molecular breeding aspects. However, in this enjoyed strategy (conventional and molecular) the target must be well defined in a complete integrated work plan. While the ability of breeders to generate large new breeding populations is almost unlimited, the evaluation and selection of these promising seedlings is the main limiting factors due to the cost and time-consuming. In this context, genomic studies at DNA level are especially useful for the development of MAS strategies. In addition, proteomic (proteins and enzymes), transcriptomic (RNA) and epigenetic (DNA Methylation and histone modifications) studies are being applied to breeding programs. Finally, these strategies at genomic, epigenomic, transcriptomic and proteomic level should be all integrated for a better understanding of the molecular mechanisms involved in the most important plant breeding aspects, which will facilitate the development and optimization of molecular markers to apply in the field exploitation of the new varieties, offering and integrating complete technological offers.
References
- Neelima R. Sinha; Plant Developmental Biology in the Post-Genomic Era. Frontiers in Plant Science 2011, 2, 11, 10.3389/fpls.2011.00011.
- National Center for Biotechnology Information (2020) NCBI Genomes. https://www.ncbi.nlm.nih.gov/genome, Accessed 12 April 2020
- Yunbi Xu; Prasanna Boddupalli; Cheng Zou; Yanli Lu; Chuanxiao Xie; Xuecai Zhang; Boddupalli M. Prasanna; Michael S. Olsen; Enhancing genetic gain in the era of molecular breeding. Journal of Experimental Botany 2017, 68, 2641-2666, 10.1093/jxb/erx135.
- Sunny Ahmar; Rafaqat Ali Gill; Ki-Hong Jung; Aroosha Faheem; Muhammad Uzair Qasim; Mustansar Mubeen; Weijun Zhou; Conventional and Molecular Techniques from Simple Breeding to Speed Breeding in Crop Plants: Recent Advances and Future Outlook. International Journal of Molecular Sciences 2020, 21, 2590, 10.3390/ijms21072590.
- Harrow J, Frankish A, González J, Taparini E; GENCODE: the reference human genome annotation for The ENCODE Project. Genome Research 2012, 22, 1760-1774, 10.1101/gr.135350.111.
- Kester Jarvis; Miranda Robertson; The noncoding universe. BMC Biology 2011, 9, 52, 10.1186/1741-7007-9-52.
- Steven Henikoff; Rapid Changes in Plant Genomes. The Plant Cell 2005, 17, 2852-2855, 10.1105/tpc.105.038000.
- Eray Tuzun; Andrew J Sharp; Jeffrey A Bailey; Rajinder Kaul; V Anne Morrison; Lisa M Pertz; Eric Haugen; Hillary Hayden; Donna Albertson; Daniel Pinkel; et al.Maynard V OlsonEvan E Eichler Fine-scale structural variation of the human genome. Nature Genetics 2005, 37, 727-732, 10.1038/ng1562.
- Pinchas Akiva; Amir Toporik; Sarit Edelheit; Yifat Peretz; Alex Diber; Ronen Shemesh; Amit Novik; Rotem Sorek; Transcription-mediated gene fusion in the human genome. Genome Research 2005, 16, 30-36, 10.1101/gr.4137606.
- Nadeau JH, & Topol FJ; The Genetics of health. Nature Genetics 2006, 38, 1095-1098.
- Gibney ER, & Nolan CM; Epigenetics and gene expression. Heredity 2010, 105, 4-13.
- Lucia Daxinger; E. Whitelaw; Transgenerational epigenetic inheritance: More questions than answers. Genome Research 2010, 20, 1623-1628, 10.1101/gr.106138.110.
- Abhinaya A; Pervasive transcription. Current Science 2011, 100, 628-630.
- Yogesh Saletore; Kate Meyer; Jonas Korlach; Igor D Vilfan; Samie Jaffrey; Christopher E. Mason; The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biology 2012, 13, 175-175, 10.1186/gb-2012-13-10-175.
- Caifu Jiang; Aziz Mithani; Eric J. Belfield; Richard Mott; Laurence D. Hurst; Nicholas P. Harberd; Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Research 2014, 24, 1821-9, 10.1101/gr.177659.114.
- Georges St. Laurent; Claes Wahlestedt; Philipp Kapranov; The Landscape of long noncoding RNA classification. Trends in Genetics 2015, 31, 239-51, 10.1016/j.tig.2015.03.007.
- Chen L, Liu P, Evans TC, & Ettwiller LM; DNA damage is a pervasive cause of sequencing errors, directly confunding variant identification. Science 2017, 355, 752-756.
- Vitaly Citovsky; Ido Keren; Casas E; Hidalgo-Carcedo C; Vavouri T; Lehner B; Klosin A; Faculty Opinions recommendation of Transgenerational transmission of environmental information in C. elegans. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017, 356, 320-323, 10.3410/f.727539298.793534731.
- Vitaly Citovsky; Ido Keren; Casas E; Hidalgo-Carcedo C; Vavouri T; Lehner B; Klosin A; Faculty Opinions recommendation of Transgenerational transmission of environmental information in C. elegans. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2017, 356, 320-323, 10.3410/f.727539298.793534731.

