 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matteo Giulietti | + 3505 word(s) | 3505 | 2021-02-01 10:09:12 | | | |
| 2 | Vivi Li | Meta information modification | 3505 | 2021-02-02 03:14:13 | | |
Video Upload Options
Extracellular vesicles (EVs) are secreted from almost all human cells and mediate intercellular communication by transferring heterogeneous molecules (i.e., DNA, RNAs, proteins, and lipids). In this way, EVs participate in various biological processes, including immune responses. Viruses can hijack EV biogenesis systems for their dissemination, while EVs from infected cells can transfer viral proteins to uninfected cells and to immune cells in order to mask the infection or to trigger a response. Several studies have highlighted the role of native or engineered EVs in the induction of B cell and CD8(+) T cell reactions against viral proteins, strongly suggesting these antigen-presenting EVs as a novel strategy for vaccine design, including the emerging COVID-19. EV-based vaccines overcome some limitations of conventional vaccines and introduce novel unique characteristics useful in vaccine design, including higher bio-safety and efficiency as antigen-presenting systems and as adjuvants.
1. Introduction
Extracellular vesicles (EVs) are small lipid particles secreted from almost all human cells types, both healthy and malignant. They can be released either directly from the plasma membrane or upon fusion among multivesicular bodies (MVBs) and the plasma membrane. Based on their size, origin, and cargo heterogeneity (i.e., DNA, proteins, various types of RNAs), EVs have been classified into several groups, such as exosomes, microvesicles, apoptotic bodies, and other vesicle types [1]. Among them, exosomes and microvesicles are very efficient mediators of cell-to-cell communication, by transferring their specific cargo to recipient cells [2][3]. For example, exosomes are involved in the delivery of genetic materials, causing epigenetic modifications in the target cells, in antigen transfer to dendritic cells (DCs) for cross-presentation to T cells, in extracellular matrix remodeling, and in several signaling pathways [2][3] (Figure 1).
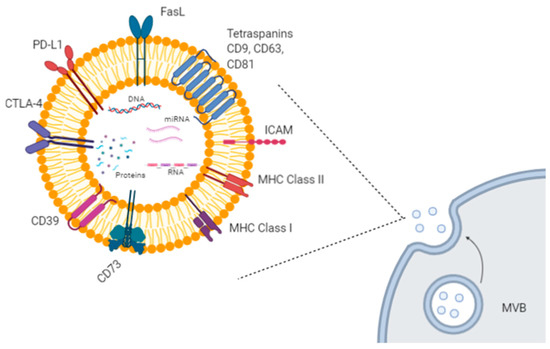
Figure 1. Exosome biogenesis and molecular cargo. Exosomes are extracellular lipid vesicles (EVs) produced within the endosomal compartment called multivesicular bodies (MVB). Exosomes’ cargo includes proteins, DNA, mRNAs, and miRNAs. Some proteins represent exosome markers (e.g., tetraspanins CD63, CD9, CD81), while other proteins are variable depending on the cell type origin, including adhesion molecules (ICAM and integrins), immune-suppressive proteins (CTLA-4, PD-L1, Fas-L, CD39, CD73), major histocompatibility (MHC) molecules, enzymes, and growth factors. Created with BioRender.com.
Here, we adhere to the nomenclature guidelines published by the International Society for Extracellular Vesicles (ISEV), which suggests the use of the general term “EVs” instead of “exosomes,” as there is still no definitive distinctive marker of each EV subtype [1].
Viruses and EVs share similar biophysical features due to their small size and similar biochemical composition, which make it difficult to separate them [4]. In addition, many enveloped viruses hijack EV biogenesis mechanisms of infected cells to enhance their dissemination by exploiting the Endosomal Sorting Complex Required for Transport (ESCRT) pathway [5]. For example, the budding of Human Immunodeficiency Virus 1 (HIV-1) at the plasma membrane [6] and the secretion of Hepatitis C Virus (HCV) from host cells require the ESCRT exosomal pathway [7][8], and even rotaviruses and noroviruses have been found within EVs [9]. As some viruses takeover EV biogenesis pathways of infected cells, there has been evolving interest in trying to understand how EV cargo is being altered during viral infections and how its transfer to surrounding uninfected cells could affect viral pathogenesis [10]. For example, HCV glycoproteins [11] and Ebola nucleoproteins [12] have been found in EVs, while an increase in human protein STING (“stimulator of IFN genes”) in CD9+ EVs was observed upon Herpes Simplex-1 (HSV-1) infection [13]. Furthermore, an altered RNA cargo of EVs released from cells infected by Respiratory Syncytial Virus (RSV) resulted in the stimulation of immune responses [14]. Therefore, EVs can both carry viral components to modulate recipient cell susceptibility to infection, and affect the host immune system to mask the infection or to trigger a response [4].
Although the viral proteins found in EVs released from infected cells and the hijacked EV biogenesis systems by viruses represent potential therapeutic targets, only few studies have assessed their contribution. Indeed, the main research field remains the use of EVs as delivery systems, as they can be easily loaded with different molecules, including drugs, antibodies, miRNAs, and siRNAs, especially in anti-tumor therapies, resulting in more specific and efficient systems than the carried molecules alone [15]. In particular, EVs showed improved stability, solubility, and biodistribution of loaded therapeutic agents [16]. For example, flotillin+/TSG101+/CD81+ EVs engineered by the addition of monoclonal HIV-1 Env antibodies on the vesicle surface and loaded with the pro-apoptotic miR-143 or the antiretroviral drug curcumin have been successfully used to destroy HIV-1-infected cells [17]. Moreover, EVs accumulate at the site with high vascular permeability, such as tumors, wounds, and sites of inflammation and of infection, due to the enhanced permeability and retention (EPR) effect. At these sites, the vasculature is leakier than healthy blood vessels, resulting in a high deposition of nanoparticles, including EVs for drug-delivery [18]. However, in general, intravenously injected EVs preferentially accumulate in the liver and spleen, probably due to the high levels of macrophages, which take up EVs and participate in the clearance of EVs [19]. In mice, inoculated EVs are rapidly cleared from the circulation, as their half-life was 2–4 min with complete clearance from blood after 4 h [20]. After roughly 30 min, the elimination phase takes over via hepatic and renal clearance, resulting in removal of intravenously injected EVs in a time span from 1 to 6 h [20][21]. Similar clearance levels and biodistribution patterns have also been observed for intravenously injected liposomes [22]. Interestingly, clearance rates and biodistribution profiles are strongly influenced by the administration route of EVs. Indeed, intraperitoneal and subcutaneous injections showed a significantly lower EV accumulation in the liver and spleen [23] and, accordingly, drug-loaded tumor-derived EVs can reach target cells at higher concentrations [22]. Notably, the route of administration is already known to also affect the pattern of metastasis (called organotropism) in mice upon inoculation of human cancer cells [24]. Therefore, clearance, bioavailability and route of administration should be taken into account during the design of both EV-based therapies (e.g., drug-loaded EVs for cancer therapies) and for EV-based vaccines.
Safety is also a key element for EV-based therapeutics and vaccines. Indeed, EV toxicity and immunogenicity have been assessed by several in vivo studies. EVs released from human mesenchymal stem cells (MSCs) did not induce genotoxic, hematological, or immunological effects during in vitro assays [25]. Human MSC-derived CD81+/CD9+/CD63+ EVs intraperitoneally injected in immunocompetent mice showed no toxicity, even in long-term expositions. Moreover, immunostimulatory effects were not observed, as lymphocyte and myeloid cell profiles and IL-6 and IFN-α levels were not altered [26]. CD81+/CD9+/CD63+ EVs from human embryonic kidney Expi293F cells did not significantly alter the transcriptome of the recipient HepG2 cells and did not induce any signs of hepatotoxicity nor proinflammatory cytokine response in BALB/c mice, after intravenous injection [27]. Unmodified CD63+/TSG101+ EVs from HEK293T cells did not elicit an immune response or toxicity in mice even after repeated intravenous and intraperitoneal administrations for 3 weeks, as observed by blood cell count and blood chemistry panels, histopathological examination, spleen immune cell composition, and evaluations of 23 circulating cytokines [28]. The Codiak BioSciences company (see Section 3 for details about its COVID-19 EV-based vaccine) engineered HEK293-derived EVs to display the anti-tumor cytokine IL-12 (exoIL-12) and, very recently, this company reported that the Phase I trial showed a favorable safety and tolerability profile [29]. It is interesting that the above studies investigated MSC- or HEK293T-derived EVs, as these EVs do not carry class I and class II major histocompatibility (MHC) proteins and B7 co-stimulatory molecules [30][31]. As these molecules are involved in stimulating the immune response, their absence in these allogeneic donor cells ensures the safety of EV vaccines. However, the use of autologous or artificial EVs may be preferable to avoid allogeneic-associated safety issues, but many allogeneic sources already fulfill good manufacturing practices and allow high EV yields, and scaled-up EV production is available. Further studies are needed to definitely ensure EV safety also in clinical settings.
2. EV-Based Vaccines: Focus on COVID-19
Since December 2019, we have been faced with the outbreak of the novel coronavirus SARS-CoV-2 (Severe Acute Respiratory Syndrome), and on 11 March 2020, the World Health Organization (WHO) declared pandemic state. The SARS-CoV-2 is a positive singe-stranded RNA virus of the Coronaviridae family, and it shares great similarity with the 2003 SARS-CoV pandemic and the Middle East Respiratory Syndrome (MERS) responsible for the outbreak in 2012 [32]. Some studies have demonstrated that the viral spike glycoprotein (S protein) facilitates the coronavirus infection of the human cells and that, in addition to the S protein, coronaviruses also need two events for cell entry: The receptor binding and the proteolytic cleavage of receptor-bound S protein that makes the spike protein active [32]. Recently, Hoffmann et al. demonstrated that such events are mediated by the human angiotensin-converting enzyme 2 (ACE2) and the Transmembrane Serine Protease 2 (TMPRSS2), respectively [33]. We have recently analyzed the effects of age, sex, diabetes, smoking habits, and pollutant on TMPRSS2 gene expression and their possible involvement in the susceptibility to viral infection and COVID-19 prognosis [34].
As the ACE2 receptor is also involved in SARS-CoV cell entry [35], it highlights the high degree of structural homology between S proteins of these Coronaviruses. However, the affinity of the SARS-CoV-2 S protein to the ACE2 is about 10–20 times higher than that of SARS-CoV, partially explaining its higher infectivity and spread [36]. Due to the structural similarity between SARS-CoV and SARS-CoV-2 spike proteins, it is interesting to highlight the results of the development of an EV-based vaccine for SARS-CoV in 2007 [37]. In particular, HSP90+/CD82+ EVs have been engineered in order to strongly enhance the spike protein loading, by creating a chimeric S protein (SGTM) as a result of the substitution of its transmembrane domain with that of the G protein of vesicular stomatitis virus (VSV). Interestingly, SGTM-containing EVs showed safety and high immunogenicity after footpad injection in mice. In particular, only two injections, instead of several needed for the S protein alone, were sufficient even without any adjuvant for the production of adequate neutralizing antibody titers. Similar results have been obtained with an adenoviral vector expressing SGTM, used as the gold standard, thus confirming the efficacy of the EV-based vaccine in inducing an immune response for SARS-CoV. Moreover, this EV-based vaccine induced higher specific antibody titers than those present in SARS patient serum [37]. Overall, this study paved the way for investigation about the EV-based vaccine also against SARS-CoV-2.
Currently, a specific antiviral treatment for COVID-19 infection is unavailable, but several clinical trials are ongoing about both new therapeutic approaches and vaccines [38][39]. The drug therapies under study include those targeting the virus infection and replication and those targeting the infected host cells and the immune system. Obviously, due to the extreme urgency, clinical trials mainly concern drug repurposing [38][39]. At the moment, different types of vaccines for SARS-CoV-2 are under study and development. Besides the classical approaches of live attenuated vaccines and inactivated virus vaccines, the most common approach is focusing on the viral Spike protein, although with different molecular strategies, such as viral-vector-based vaccines, mRNA vaccines, or those with the full-length S protein or its subunit (receptor binding domain (RBD)) [38][39]. Recently, BNT162b2 (Pfizer, New York City, USA and BioNTech, Mainz, Germany) and mRNA-1273 (Moderna, Cambridge, MA, USA) vaccines have been approved by the medicine regulatory authorities of the UK, USA, and EU, and a mass vaccination campaign started in December, whereas ChAdOx1 nCoV-19 (AstraZeneca, Cambridge, UK and Oxford University, UK) has been very recently approved by UK authority.
Additionally, in the last months, vaccines based on EVs or exosome-like vesicles for COVID-19 have been under development by some biotech companies (Figure 3).
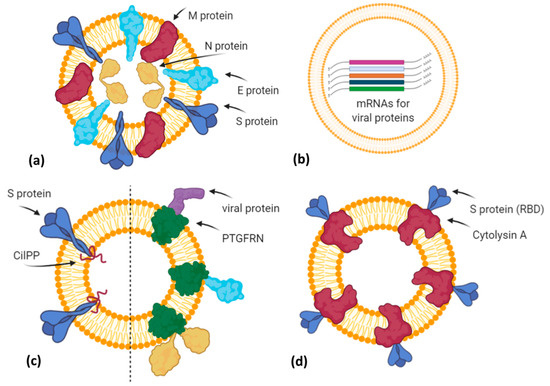
Figure 3. A simplified graphical representation of different EV-based vaccine strategies under development by various biotech companies. (a) Capricor Therapeutics transfected HEK293 cells with vectors for Spike, Nucleocapsid, Membrane, and Envelope SARS-CoV-2 proteins in order to release EVs (or virus-like particles) carrying viral antigens in their native conformation; (b) Carpicor Therapeutics formulated a COVID-19 vaccine by loading into EVs mRNAs for full-length S protein and for modified S, N, M, and E proteins inserted within Lamp1 protein for a better presentation on MHC I and II molecules. Allele Biotechnology and Pharmaceuticals is also developing an EV-based mRNA vaccine, but details are not available; (c) CoVEVax vaccine by Ciloa company consists of EVs displaying full S protein, thanks to the EV-sorting peptide CilPP (left). Similarly, exoVACC™ vaccine of Codiak BioSciences consists of EVs engineered to display still-undeclared SARS-CoV-2 proteins, thanks to the scaffold protein PTGFRN (right); (d) bacterial Outer Membrane Vesicles (OMVs) engineered by Versatope Therapeutics for the display of Spike protein (receptor binding domain (RBD) portion) by fusing it with the OMV-anchoring protein cytolysin A (ClyA). Created with BioRender.com.
The company Capricor Therapeutics [40] is also working on its EV platform technology as a potential COVID-19 vaccine. Notably, Capricor is developing two different EV-based vaccines designed to stimulate a long-lasting protective immune response to SARS-CoV-2. The first type is an EV display vaccine consisting of human HEK293 cells transfected with vectors for the expression of the four structural SARS-CoV-2 proteins (Spike, Nucleocapsid, Membrane, Envelope). In turn, the released EVs will carry all viral antigens in their native context and conformation (Figure 3a). Previously, it has been demonstrated that immunization with multiple protein forms allows the modulation of the magnitude and the nature of the immune response, in terms of cytokine production and Th1 or Th2 stimulation [41]. However, regarding this vaccine candidate, further details are not available. The second type is an mRNA vaccine formulated by EVs loaded with five different mRNAs coding for modified SARS-CoV-2 Spike, Nucleocapsid, Membrane, and Envelope proteins (LSNME) and for the full-length Spike of Wuhan-1 isolate (SW1). In November 2020, Capricor and Johns Hopkins University researchers published, as a pre-print version, encouraging results of pre-clinical trials for their multivalent EV-based mRNA vaccine [42]. They have combined the EV-based mRNA delivery and viral antigen expression compatible for antigen presentation by MHC Class I and II molecules. In particular, they designed mRNAs coding for RBD (receptor binding domain) of the S protein, full-length N protein, and soluble fragments of M and E proteins, but expressed within the extracellular domain of Lamp1 human protein, which is known to be subjected to degradation into short peptides for antigen presentation by the MHC I pathway and, in antigen-presenting cells (APCs), by the MHC II pathway (Figure 3b). HEK293 CD9+/CD63+ EVs loaded with LSNME and functional SW1 mRNAs (LSNME/SW1 vaccine) were injected intramuscularly into C57BL/6J mice at various concentrations. After the first injection, animals received two further boosters after 3 and 6 weeks, respectively. ELISA assays showed a concentration-dependent antibody response for both N and S proteins, and developed immunity lasting up to 2 months after the second boost injection. Furthermore, the LSNME/SW1 vaccine caused a substantial increase in CD4+ and CD8+ T-cells that proliferated upon addition of N and S recombinant proteins to the culture medium of splenocytes, thus confirming that this vaccine formulation is also able to induce cellular immune responses. In particular, S-induced T-cells showed high expression of IFN-γ (Th1 response) but low levels of IL4 (Th2 response). Finally, mice did not show vaccine-induced adverse reactions, such as injection site inflammation, altered body growth, organ morphology, or blood cell profiles.
The Ciloa company developed a COVID-19 vaccine, called CoVEVax, based on HEK293T-derived CD81+/CD63+/CD9+ EVs. These EVs have been engineered to display the full S protein on their surface, thanks to the fusion with the patented EV-sorting peptide CilPP (Figure 3c). In addition, the S protein has been stabilized by the substitution of two consecutive prolines (K986P, V987P). In a pre-print article, the preclinical results about safety and efficacy have been described [43]. In particular, mice were injected subcutaneously, without adjuvants, with the two components of this vaccine, i.e., the DNA vector for the engineered EVs and the HEK293T-derived engineered EVs. Indeed, only this combination elicited both a humoral and cellular response, measured as levels of specific IgG to S1 or S2 peptide and as antigen-specific IFN-γ production.
Another biotech company, the Codiak BioSciences [44], in collaboration with the Ragon Institute of MGH, MIT, and Harvard University, is studying the potential of its exoVACC™ vaccine platform for SARS-CoV-2. In particular, exoVACC™ is a modular vaccine system that exploits the unique EV properties, including the simultaneous delivery of specific antigens and immuno-stimulatory adjuvants to the antigen-presenting cells (APCs), for the stimulation of the innate cellular and humoral immune reaction. It is based on the proprietary engEx™ platform for the EV surface display, which utilizes the scaffold protein PTGFRN (Prostaglandin F2 receptor negative regulator), known to preferentially sort in EVs. It is a single-pass transmembrane glycoprotein that enables the high-density surface display of fused proteins of interest, including cytokines, antibody fragments, and other immunomodulatory proteins or specific antigens up to 170 kDa. Due to its high abundance in EVs, PTGFRN showed better packaging and antigen display efficiencies than conventional scaffold proteins, such as CD81, LAMP2B, and the vector system called “pDisplay” based on platelet-derived growth factor receptor (PDGFR) [45]. However, exoVACC™ is still in the research phase, as multiple combinations of SARS-CoV-2 antigens and adjuvants can be produced, and their effectiveness and specificity in vitro and in animal models should be assessed (Figure 3c).
The company Allele Biotechnology and Pharmaceuticals [46] has recently announced the development of an iPSC (induced pluripotent stem cell) line transfected with different mRNAs encoding the SARS-CoV-2 antigen proteins (Figure 3b). These cells can produce a high amount of EVs carrying both viral mRNAs and the corresponding proteins. Allele states that this system overcomes two issues: (i) A vaccine with multiple mRNAs and proteins could have better performances than those with a single mRNA, like Pfizer/BioNTech and Moderna vaccines or those in the ongoing trials; (ii) the Pfizer/BioNTech vaccine needs to be stored at –80 °C, but as EVs protect mRNA from degradation, Allele’s iPSC-derived EVs resulted to be intact for months even when stored at 4 °C. However, similarly to Codiak, no results have been published, nor further methodological details declared.
Finally, Versatope Therapeutics [47] is developing a vaccine against SARS-CoV-2 based on exosome-like nano-sized vesicles called Outer Membrane Vesicles (OMVs). OMVs are lipid vesicles naturally produced by bacteria and present similar characteristics to human EVs. OMVs have been engineered by Versatope in order to display the RBD portion of the Spike protein thanks to its fusion with the OMV-anchoring protein cytolysin A (ClyA), similarly to their candidate vaccine against influenza A virus [48][49] (Figure 3d). In previous studies, OMVs have also been exploited as drug delivery vehicles, cancer immunotherapy agents, immune adjuvants, and vaccines against their parent bacteria [50] or engineered to express antigens of interest from influenza A H1N1 Virus and MERS-CoV [51], in order to elicit protection mediated by antigen cross-presentation to CD8(+) T cells [52]. The concerns about OMV biosafety, due to their bacterial origin and consequent possible inflammatory response, seem to be solved by chemical and genetic approaches that efficiently reduce OMV reactogenicity in humans. In particular, the use of nonionic detergents and chelating agents causes the reduction or dissolvement of OMV lipopolysaccharides (LPS) after the isolation of these bacterial vesicles, whereas the genetic engineering of genes involved in the biosynthetic pathway of LPS allows for the production of recombinant OMVs with attenuated LPS forms [53].
In case these EV-based vaccines demonstrate lower immune responses, it could be useful to consider the use of the modified mRNA coding for the prefusion conformation of the Spike protein, as employed so far by Moderna and Pfizer/BioNTech. This modified mRNA could be loaded directly into extracellular vesicles or into cells by transfection in order to produce vesicles carrying this mRNA. The Spike protein undergoes structural rearrangements in order to fuse with the host cell membrane, from an unstable prefusion conformation to a highly stable post-fusion conformation. It has been demonstrated that stabilization of prefusion-immunogens, preserving epitopes subjected to neutralization, is a promising vaccine strategy for enveloped viruses [54]. Previously, it has been described as a successful stabilization of MERS-CoV and SARS-CoV in prefusion conformation by 2 proline substitutions (2P) in the central helix and heptad repeat 1 of the Spike protein [55]. This mutation is responsible for higher immunogenicity of the MERS-CoV S(2P) protein than wild-type protein, and better stability of the S protein of other betacoronaviruses. This finding indicated a possible strategy for development of vaccines against betacoronaviruses, including SARS-CoV-2. Indeed, the company Moderna substituted the two prolines in SARS-CoV-2 S protein residues 986 and 987 in order to obtain prefusion-stabilized protein and shortly after started the production of mRNA-LNP for SARS-CoV-2 S(2P) protein (mRNA-1273) [54]. Similarly, Pfizer/BioNTech also based their vaccines on mRNA coding for S protein (2P), but with additional modifications. In particular, their vaccine is formulated as a lipid-nanoparticle loaded with N-methyl-pseudouridine (m1Ψ) nucleoside-modified mRNA coding for S protein (2P) containing a native furin cleavage site resulting in two cleavage fragments. The methyl-pseudouridine modification and the optimization of noncoding elements are responsible for enhanced in vitro RNA translation and reduced immune sensing [56]. Previously, m1Ψ-modified mRNA vaccines have been found immunogenic for other viruses, including Zika and HIV-1 [57][58]. Finally, as this recombinant trimeric S (2P) protein is still able to bind the human ACE2 receptor and human anti-RBD antibodies with high affinity, it proves its structural and functional integrity [56].
References
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383.
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228.
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572.
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820.
- Lorizate, M.; Sachsenheimer, T.; Glass, B.; Habermann, A.; Gerl, M.J.; Krausslich, H.G.; Brugger, B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell Microbiol. 2013, 15, 292–304.
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113.
- Tamai, K.; Shiina, M.; Tanaka, N.; Nakano, T.; Yamamoto, A.; Kondo, Y.; Kakazu, E.; Inoue, J.; Fukushima, K.; Sano, K.; et al. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 2012, 422, 377–385.
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.L.; Mutsafi, Y.; De Jesus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220.
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. Cloaked Viruses and Viral Factors in Cutting Edge Exosome-Based Therapies. Front. Cell Dev. Biol. 2020, 8, 376.
- Zicari, S.; Arakelyan, A.; Palomino, R.A.N.; Fitzgerald, W.; Vanpouille, C.; Lebedeva, A.; Schmitt, A.; Bomsel, M.; Britt, W.; Margolis, L. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology 2018, 524, 97–105.
- Pleet, M.L.; Mathiesen, A.; DeMarino, C.; Akpamagbo, Y.A.; Barclay, R.A.; Schwab, A.; Iordanskiy, S.; Sampey, G.C.; Lepene, B.; Nekhai, S.; et al. Ebola VP40 in Exosomes Can Cause Immune Cell Dysfunction. Front. Microbiol 2016, 7, 1765.
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996.
- Chahar, H.S.; Corsello, T.; Kudlicki, A.S.; Komaravelli, N.; Casola, A. Respiratory Syncytial Virus Infection Changes Cargo Composition of Exosome Released from Airway Epithelial Cells. Sci. Rep. 2018, 8, 387.
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as drug carriers for cancer therapy and challenges regarding exosome uptake. Biomed. Pharmacother. 2020, 128, 110237.
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614.
- Zou, X.; Yuan, M.; Zhang, T.; Wei, H.; Xu, S.; Jiang, N.; Zheng, N.; Wu, Z. Extracellular vesicles expressing a single-chain variable fragment of an HIV-1 specific antibody selectively target Env(+) tissues. Theranostics 2019, 9, 5657–5671.
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017.
- Imai, T.; Takahashi, Y.; Nishikawa, M.; Kato, K.; Morishita, M.; Yamashita, T.; Matsumoto, A.; Charoenviriyakul, C.; Takakura, Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracel. Vesicles 2015, 4, 26238.
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84.
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494.
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control Release 2015, 199, 145–155.
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316.
- Giulietti, M.; Bastianoni, M.; Cecati, M.; Ruzzo, A.; Bracci, M.; Malavolta, M.; Piacenza, F.; Giacconi, R.; Piva, F. MetaTropismDB: A database of organ-specific metastasis induced by human cancer cell lines in mouse models. Database 2020.
- Maji, S.; Yan, I.K.; Parasramka, M.; Mohankumar, S.; Matsuda, A.; Patel, T. In vitro toxicology studies of extracellular vesicles. J. Appl. Toxicol. 2017, 37, 310–318.
- Mendt, M.; Kamerakar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263.
- Saleh, A.F.; Lazaro-Ibanez, E.; Forsgard, M.A.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001.
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; La Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730.
- Lewis, N.D.; Sia, C.L.; Kirwin, K.; Haupt, S.; Mahimkar, G.; Zi, T.; Xu, K.; Dooley, K.; Jang, S.C.; Choi, B.; et al. Exosome surface display of IL-12 results in tumor-retained pharmacology with superior potency and limited systemic exposure compared to recombinant IL-12. Mol. Cancer Ther. 2020.
- Yi, Y.W.; Lee, J.H.; Kim, S.Y.; Pack, C.G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21, 665.
- Li, J.; Chen, X.; Yi, J.; Liu, Y.; Li, D.; Wang, J.; Hou, D.; Jiang, X.; Zhang, J.; Wang, J.; et al. Identification and Characterization of 293T Cell-Derived Exosomes by Profiling the Protein, mRNA and MicroRNA Components. PLoS ONE 2016, 11, e0163043.
- Yang, Y.; Peng, F.; Wang, R.; Yange, M.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun 2020, 109, 102434.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Piva, F.; Sabanovic, B.; Cecati, M.; Giulietti, M. Expression and co-expression analyses of TMPRSS2, a key element in COVID-19. Eur. J. Clin. Microbiol. Infect. Dis. 2020.
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263.
- Kuate, S.; Cinatl, J.; Doerr, H.W.; Uberla, K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology 2007, 362, 26–37.
- Ghaebi, M.; Osali, A.; Valizadeh, H.; Roshangar, L.; Ahmadi, M. Vaccine development and therapeutic design for 2019-nCoV/SARS-CoV-2: Challenges and chances. J. Cell Physiol. 2020.
- Omolo, C.A.; Soni, N.; Fasiku, V.O.; Mackraj, I.; Govender, T. Update on therapeutic approaches and emerging therapies for SARS-CoV-2 virus. Eur. J. Pharmacol. 2020, 173348.
- Capricor Therapeutics. Available online: https://capricor.com/covid-19/ (accessed on 26 January 2021).
- Morel, P.A.; Falkner, D.; Plowey, J.; Larregina, A.T.; Falo, L.D. DNA immunisation: Altering the cellular localisation of expressed protein and the immunisation route allows manipulation of the immune response. Vaccine 2004, 22, 447–456.
- Tsai, S.; Guo, C.; Atai, N.A.; Gould, S.J. Exosome-Mediated mRNA Delivery For SARS-CoV-2 Vaccination. bioRxiv 2020.
- Polak, K.; Greze, N.; Lachat, M.; Merle, D.; Chiumento, S.; Bertrand-Gaday, C.; Trentin, B.; Mamoun, R.Z. Extracellular vesicle-based vaccine platform displaying native viral envelope proteins elicits a robust anti-SARS-CoV-2 response in mice. bioRxiv 2020.
- Codiak BioSciences. The exoVACC Vaccine Platform for SARS-CoV-2. Available online: https://ir.codiakbio.com/news-releases/news-release-details/codiak-biosciences-collaborates-ragon-institute-evaluate (accessed on 26 January 2021).
- Dooley, K.; McConnell, R.E.; Xu, K.; Lewis, N.D.; Haupt, S.; Youniss, M.R.; Martin, S.; Sia, C.L.; McCoy, C.; Moniz, R.J.; et al. A Versatile Platform for Generating Engineered Extracellular Vesicles with Defined Therapeutic Properties. Mol. Therapy 2020.
- Allele Biotechnology and Pharmaceuticals Inc. COVID-19. Available online: https://www.allelebiotech.com/covid19 (accessed on 26 January 2021).
- Versatope Therapeutics, Inc. Available online: https://www.versatope.com/ (accessed on 26 January 2021).
- Rappazzo, C.G.; Watkins, H.C.; Guarino, C.M.; Chau, A.; Lopez, J.L.; DeLisa, M.P.; Leifer, C.A.; Whittaker, G.R.; Putnam, D. Recombinant M2e outer membrane vesicle vaccines protect against lethal influenza A challenge in BALB/c mice. Vaccine 2016, 34, 1252–1258.
- Watkins, H.C.; Rappazzo, C.G.; Higgins, J.S.; Sun, X.; Brock, N.; Chau, A.; Misra, A.; Cannizzo, J.P.B.; King, M.R.; Maines, T.R.; et al. Safe Recombinant Outer Membrane Vesicles that Display M2e Elicit Heterologous Influenza Protection. Mol. Ther. 2017, 25, 989–1002.
- Li, M.; Zhou, H.; Yang, C.; Wu, Y.; Zhou, X.; Liu, H.; Wang, Y. Bacterial outer membrane vesicles as a platform for biomedical applications: An update. J. Control. Release 2020, 323, 253–268.
- Shehata, M.M.; Mostafa, A.; Teubner, L.; Mahmoud, S.H.; Kandeil, A.; Elshesheny, R.; Frantz, R.; La Pietra, L.; Pleschka, S.; Osman, A.; et al. Bacterial Outer Membrane Vesicles (OMVs)-based Dual Vaccine for Influenza A H1N1 Virus and MERS-CoV. Vaccines 2019, 7, 46.
- Schetters, S.T.T.; Jong, W.S.P.; Horrevorts, S.K.; Kruijssen, L.J.W.; Engels, S.; Stolk, D.; Daleke-Schermerhorn, M.H.; Garcia-Vallejo, J.; Houben, D.; Unger, W.W.J.; et al. Outer membrane vesicles engineered to express membrane-bound antigen program dendritic cells for cross-presentation to CD8(+) T cells. Acta Biomater. 2019, 91, 248–257.
- Rossi, O.; Citiulo, F.; Mancini, F. Outer membrane vesicles: Moving within the intricate labyrinth of assays that can predict risks of reactogenicity in humans. Hum. Vaccin Immunother. 2020, 1–13.
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571.
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357.
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. bioRxiv 2020.
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251.
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15, 36–47.

