 +1 credit
+1 credit
Video Upload Options
WASF3 (WAVE3), a Wiskott–Aldrich syndrome protein family member, appears to play a major role not only in the regulation of actin cytoskeleton dynamics but also in cancer cell invasion/metastasis. Blocking the WASF3-dependent metastatic signaling network remains an attractive and promising therapeutic option for the treatment of advanced tumors.
1. Introduction
The dispersal of cancer cells from their primary growth site to secondary growth sites in the body represents the metastatic process and the leading cause of death in cancer patients today [1]. Despite their clinical implications, the genes and mechanisms underlying this process have yet to be fully elucidated owing to their expansive influence and inherent complexity. Identifying and characterizing the lead drivers of metastasis has, therefore, remained the primary motive for many cancer research groups, including ours.
One family of proteins, the Wiskott-Aldrich syndrome protein (WASP) family, plays an important role in cell invasion and metastasis through the activation of leading edge membrane structures. The family consists of five members that are divided into two subfamilies based on their structural similarities. The WASP subfamily consists of WASP and N-WASP; and the WASF (also referred to as WAVE) subfamily consists of WASF1, WASF2, and WASF3 [2]. For all members, phosphoactivation following extracellular stimulus (e.g., cytokines or growth factors) leads to the exposure of their C-terminus motifs which then interact with many factors, including the actin-related protein (Arp) 2/3 complex [2].
In particular, WASF members mediate the activation of Arp2/3 through a five-subunit WASF regulatory complex (WRC) which acts downstream of the GTPase Rho family member Rac [3][4]. In their normal inactive state, WASF proteins maintain an autoinhibited conformation through interactions between their N- and C-termini formed by WRC binding components, functionally blocking the verprolin-cofilin-acidic (VCA) region where the Arp2/3 complex binds (Figure 1) [5]. Following activation by Rac through constituents like the cytoplasmic FMR1 interacting protein 1 (CYFIP1, also referred to as SRA1), however, a conformational change exposes the VCA region of WASFs, allowing the CA domain to associate with the Arp2/3 complex and the V domain with monomeric actin [5]. The Arp2/3 complex then initiates actin polymerization and branched filament network formation by generating a new nucleation core and binding to pre-existing filaments [6]. From this branched nucleation process, actin projections at the leading edge of the cell (lamellipodia) and actin matrix-degrading structures (invadopodia) can be subsequently formed and used to facilitate cell motility and matrix remodeling [5]. Under normal conditions, the formation of these structures is strictly governed to safeguard tissues against disruption and damage. Under pathological conditions, however, cancer cells exploit this process to locally invade and metastasize throughout the body.
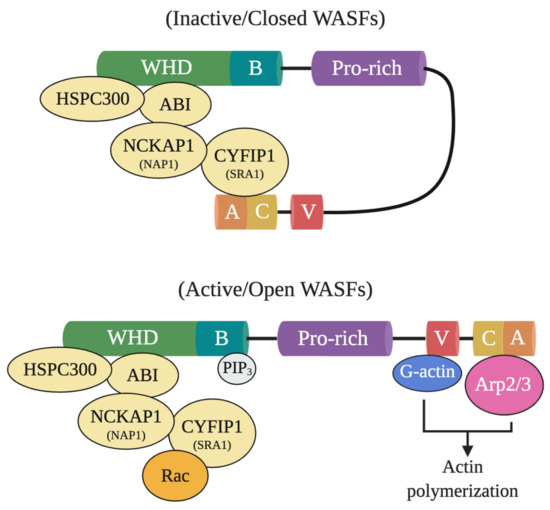
Figure 1. Inactive and active WASF domain confirmations. At resting state, WASFs exist in an autoinhibited closed conformational state due to the interactions between members of the N-terminally located WASF homology domain (WHD) and the C-terminus. Members of the protein complex at WHD include HSPC300, ABI, NCKAP1 (NAP1), CYFIP1 (SRA1), and WASF1/2/3. Upon activation by Rac, WHD interactions are disrupted and WASFs adopts an open conformation where their verprolin-cofilin-acidic (VCA) region can interact with the actin-related protein (Arp) 2/3 complex and monomeric actin (G-actin). Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) may also recruit the WASF complex to the membrane by binding to the basic domain (B) of WASFs.
Although each of the WASP and WASF proteins form networks of regulatory steps, of them, WASF3 represents one of the most central and critical intermediates in the signaling pathways promoting metastasis. It should be noted, however, that while WASF1 and WASF2 are not essential to invadopodium formation [7], WASF2, in particular, has attracted significant attention for its potential as a prognostic indicator and role in the invasion of cancers, like melanoma [8], breast [9], and pancreatic [10] cancers.
In normal cells, WASF3 serves in the transduction of signals leading to changes in cell morphology and cytoskeletal organization through the aforementioned regulatory mechanism and is predominately expressed in the nervous system [11]. In various human cancers, like colon [12], prostate [13], and pancreatic [14], however, WASF3 is upregulated and, in the case of breast cancer, linked to increased invasive and metastatic cell potential through the regulation of epithelial-to-mesenchymal transition (EMT) [15][16]. Moreover, the expression of WASF3 has been reported to be positively correlated with poorer prognosis in gastric cancer [17], non-small cell lung cancer [12], and hepatocellular carcinoma patients [18] and to be enriched in triple-negative breast cancer stem cells where it promotes self-renewal and may contribute to chemoresistance [19]. Additionally, the protein has been found to promote the invasion of ovarian cancer cells and to play an important role in their secretion of the pluripotent transcription factors Oct4 and SOX2 [20]. Lastly, WASF3 has also been linked to the regulation of matrix metalloproteinase (MMP) production, providing invading cells a means to remodel the extracellular matrix [21].
2. Genetic Dissection of WASF3-Related Signal Transduction Pathways
2.1. Regulation of WASF3 Transcription
2.1.1. Regulation by Signal Transducer and Activator of Transcription 3 (STAT3)
Little had been revealed regarding the regulation of WASF3 at the transcription level, however, our experiments have shown that there are three potential binding sites for STAT3 within 1100 base pairs upstream of the WASF3 transcription initiation site [22]. These sites are located at the positions -894 to -886, -915 to -906, and -926 to -919. Interleukin 6 (IL-6) is one of the many cytokines used by tumor cells to facilitate invasion and metastasis. Our results showed that within 2 min of application, IL-6 can transition WASF3 from its inactive, non-phosphorylated form in the cytoplasm to its active, phosphorylated form, which relocates to the cytoplasmic membrane and engages with Janus kinase 2 (JAK2) (Figure 2). An increase in WASF3 gene transcription is seen to follow. Specifically, STAT3 is phosphoactivated by JAK2 kinase and relocates to the nucleus to bind to the previously mentioned binding sites to increase the transcription of WASF3 [22]. Inhibiting the activation of STAT3 by its inhibitor S3I-201 leads to reduced WASF3 levels, even with the application of IL-6. Although there are many receptors, this activation process was found to be largely achieved through the binding of IL-6 to the GP130 receptor. Upon blocking GP130 with BR-3 monoclonal antibody, WASF3 levels showed no change in the presence of IL-6. It was also found that JAK2 phosphoactivated WASF3 protein and that inactivation of JAK2 by the JAK kinase inhibitor AG490 led to a reduction in activated WASF3 levels [22].
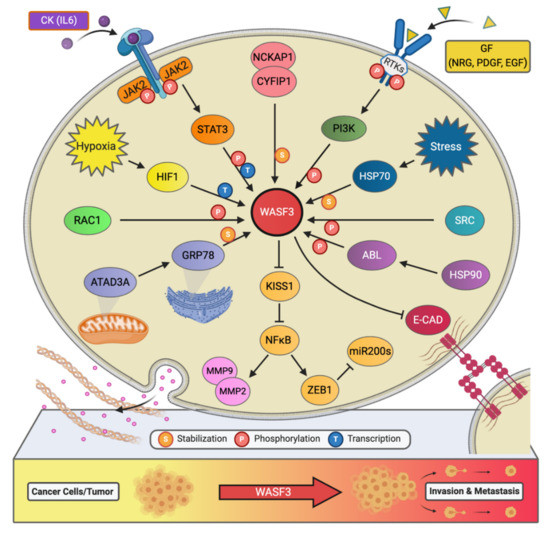
Figure 2. The signaling network of WASF3 in cancer metastasis. The WASF3-dependent signaling pathways and related regulatory networks critical to controlling cancer metastasis are summarized here.
2.1.2. Regulation by Hypoxia-Inducible Factor 1-Alpha (HIF1A)
It is well known that hypoxia affects the ability of cancer cells to invade and metastasize through changes in molecular pathways. Constituents of these pathways, therefore, represent potential therapeutic targets for preventing metastasis. In particular, WASF3 is a hypoxia-inducible gene, meaning that it contains at least one or more hypoxia responsive elements (HRE) which act as binding sites for transcription factors [23]. The levels of WASF3 protein increased significantly in MDA-MB-231 and MCF7 breast cancer cells incubated under hypoxic conditions. In the promoter region of the WASF3 gene, four HREs were identified. HRE1, 2, and 3 are located in between -27 and -79 base pairs of the first exon of the WASF3 gene, and HRE4 is located distally around -721 and -725 base pairs to the same reference point. HIF1s bind to these HRE and increase the transcription of WASF3 in a hypoxic environment. The HIF1A factor is required to facilitate this increment change in WASF3 levels, as knockdown of HIF1A, but not HIF2A, was found to reduce the levels of WASF3 in cells undergoing hypoxia (Figure 2) [22]. The phosphoactivation process of WASF3 was also increased under hypoxic conditions, as well as the levels of MMP1, MMP3, and MMP9. One study showed that combining anti-HIF (YC-1) and anti-VEGF (SU5416) treatments in MDA-MB-231 cells under these conditions decreases WASF3 levels dramatically in comparison to anti-VEGF treatment alone [22]. The possible mechanism behind this observation is that inhibition of VEGF may target the angiogenic ability of cancer cells and create a hypoxic environment that, in turn, increases WASF3 levels. The use of conventional anti-angiogenic cancer treatments that rely solely on this type of medication, therefore, might need to be reconsidered.
2.2. Regulation of WASF3 Protein Stability
2.2.1. Regulation by Heat Shock Protein 70 (HSP70)
Chaperone proteins play critical roles in facilitating client protein activation and/or maintaining their stability. Examples of chaperone proteins include HSP90 and HSP70. While HSP90 has been linked to WASF3 activation through ABL kinase, HSP70 has been seen to preserve WASF3 protein stability in the cytoplasm and to protect it from proteasome degradation [24]. Using MDA-MB-231 cells expressing a series of truncated, HA-tagged WASF3 constructs, HSP70 interaction with WASF3 was found to take place at WASF3’s N-terminus (Figure 2). Interfering with the transcriptional activation of HSP70 with KNK437 in HEK-293 cells also led to a reduction in the protein levels of both HSP70 and WASF3 while leaving WASF3 mRNA levels unchanged. Accordingly, heat shock experiments across various cancer cell lines further resulted in increased levels of HSP70 and WASF3 protein without an increase in WASF3 mRNA [24].
Given that HSP70 binding frequently functions to target client proteins for degradation, treatment with the proteasome inhibitor MG132 alone or in combination with HSP70 inhibitors provided further clarification into the HSP70-mediated stabilization of WASF3. Specifically, it was found that disrupting the association of HSP70 and WASF3 using PEP, even in the presence of MG132, led to a reduction in WASF3 protein levels [24]. This suggests that it is the formation of the HSP70-WASF3 complex, rather than endogenous HSP70 protection from proteasome degradation that provides WASF3 stability. Further study also revealed that silencing or pharmacological inhibition of HSP70 hinders cell migration while overexpressing it elevates WASF3 protein levels and invasion [24]. Aside, others have reported that an HSP70/WASF3/MMP9 axis can be further upregulated to promote breast cancer cell invasion through p63α, a member of the p53 family that is critical to HSP70 expression [25]. Taken together, these findings contribute to the basis of our understanding of HSP70’s importance in WASF3 stabilization and, in turn, WASF3-mediated cancer cell invasion and metastasis.
2.2.2. Regulation by ATAD3A-Dependent GRP78
AAA domain-containing 3A (ATAD3A) is a ubiquitously expressed mitochondrial membrane ATPase that contributes to the regulation of mitochondrial dynamics [26]. Recently, ATAD3A has been the subject of intense interrogation for its role throughout cancer progression and, more specifically, its role in regulating WASF3 facilitated metastasis [27][28]. In engraftment experiments using MDA-MB-231 cells and immunocompromised mice, knockdown of ATAD3A markedly suppressed tumor growth, neovascularization, and the incidence of metastatic colonies (Figure 2). In vitro, ATAD3A silencing was also found to significantly reduce WASF3 protein levels while leaving WASF3 mRNA levels unchanged. Importantly, this silencing was additionally accompanied by a decrease in cell anchorage-independent growth and invasion [28].
While HSP70 serves to stabilize WASF3 in the cytoplasm, it is presumed that ATAD3A, alongside GRP78, maintain this role in the mitochondria. In particular, inactivation of HSP70 was seen to significantly reduce the levels of cytoplasmic WASF3 but not the levels of mitochondrial-associated WASF3. Immunofluorescent signaling representing WASF3-mitochondria association, however, was seen to be entirely diminished following ATAD3A knockdown. Through ATAD3A truncation experiments, it was additionally observed that WASF3 interacts exclusively with the N-terminus of ATAD3A and that this interaction is not only crucial to WASF3 stabilization but independent of HSP70. Aside, the N-terminus of WASF3 was found to penetrate the outer mitochondrial membrane, which protects against proteolysis. Interestingly, the endoplasmic reticulum (ER) resident chaperone protein GRP78 was also found to be implicated in ATAD3A-mediated WASF3 stabilization (Figure 2). Under normal conditions, GRP78 contributes to the coordination of the unfolded protein response following ER stress [28].
In both breast and colon cancer cell lines, it was observed that while ATAD3A knockdown decreased the protein levels of GRP78 and WASF3, WASF3 levels could be ameliorated by overexpressing GRP78. Moreover, in cells overexpressing WASF3, the half-life of WASF3 could be significantly extended if GRP78 was also overexpressed. Accordingly, GRP78 knockdown did not affect either the protein levels of ATADA3 or gene expression of WASF3 but did reduce the levels of WASF3 protein. Lastly, ATAD3A-GRP78 binding was also observed between ER and mitochondrial membranes and, under ER stress, it was seen that WASF3 stabilization was enhanced through increased WASF3-GRP78 interaction [28]. ATAD3A stabilization of WASF3, therefore, is likely dependent on GRP78. Further study in our group reveals that ATAD3A suppresses CDH1/E-cadherin expression through its regulation of WASF3 that is associated with GRP78 function [28].
2.3. Regulation of WASF3 Protein Phosphorylation
2.3.1. Regulation by Phosphoinositide 3-Kinase (PI3K)
In addition to IL-6, platelet-derived growth factor (PDGF) has also been found to influence the activity of the WASF3 protein [29]. In particular, following MDA-MB-231 cell treatment with PDGF, it was discovered that interaction between WASF3 and PI3K is required for lamellipodia formation and cell migration. Structurally, the PI3K protein consists of two subunits, a p110 catalytic subunit and a p85 regulatory subunit which is an important effector for actin cytoskeleton remodeling and, thus, cell migration. Upon PDGF binding, PI3K is recruited to the activated PDGFR and binds to it through its p85 C-terminus SRC homology 2 (SH2) domain, resulting in internalization of the complex into the cytoplasm. Interestingly, it was found that WASF3 also interacts with PI3K at this same SH2 domain and that treating cells with the PI3K inhibitor LY294002 significantly decreases lamellipodia formation and cell migration, suggesting that WASF3-p85 interaction may play a part in actin polymerization and cell invasion (Figure 2) [29].
In another study, pharmacological inhibition of PI3K isoforms was similarly found to decrease cell motility, as well as suppress WASF3 induction in TLR5/7-treated ovarian SK-OV-3 cancer cells [20]. It was also revealed that following the PI3K-mediated activation of WASF3, WASF3 goes on to play a critical role in the expression of mesothelin and production of Oct4/SOX2 in TLR5/7-mediated SK-OV-3 cell invasion [20]. Aside, it has recently been reported that WASF3 phosphorylation acts to modulate a positive feedback loop between WASF3 and the PI3K-TGF-β-EGF signaling pathways [16], which supports previous findings that depletion of WASF3 expression in breast cancer cells prevents TGF-β-mediated EMT and lamellipodia formation in metastatic cells [30].
2.3.2. Regulation by JAK2
Interestingly, an increase in phosphoactivated WASF3 and WASF3 protein levels was observed in prostate cancer DU145 cells following the treatment of IL-6 [22]. The same tendency was also seen in breast cancer MDA-MB-231 cells after IL-6 stimulation [22]. The cells receiving the pan JAK inhibitor AG490 showed no activation of WASF3, while those receiving the STAT3 inhibitor S31-201 showed reduced levels of activated WASF3 owing to a decrease in the total amount of WASF3 [22]. To dig deeper into which of the JAK members were implicated, we knocked down either JAK1 or JAK2 in MDA-MB-231 cells and subsequently found a significant reduction in WASF3 levels in JAK2 knockdown cells but not JAK1 [22]. From these findings, we concluded that IL-6 induces WASF3 phosphoactivation through the JAK2 pathway, which facilitates WASF3’s relocation to the cell membrane and the reorganization of the intracellular actin cytoskeleton, leading to cell migration and metastasis.
2.3.3. Regulation by Abelson Tyrosine Kinase (ABL)
Another critical component to WASF3’s phosphoactivation is ABL, whose presence in the WASF3 immunocomplex was confirmed through immunoprecipitation (IP) analysis of MDA-MB-231 cells treated with PDGF. In one study, treatment of MDA-MB-231 cells with the ABL kinase inhibitor Gleevec (STI-571) led to a dramatic decrease in WASF3 phosphorylation levels [11]. Moreover, four tyrosine residues in the amino acid sequence of WASF3 were identified as possible targets of ABL mediated phosphorylation, including Tyr-151, Tyr-248, Tyr-337, and Tyr-486. Only when all four of these residues are mutated does ABL kinase lose its ability to carry out its role in this phosphorylation process and lamellipodia formation and cell invasion decrease [11].
2.3.4. Regulation by HSP90
Chaperone proteins like HSP90 are necessary for the proper folding and degradation of client proteins, affecting their stability and levels inside the cell. When PC3, MDA-MB-231, SKBR3, and COS7 cell lines were treated with the HSP90 inhibitor 17-AAG that acts by binding to the ATP/ADP binding pocket on HSP90’s N-terminus, there were no observable changes in WASF3 protein levels [24]. Similar results were also seen with the treatment of Novobiocin, another HSP90 inhibitor that instead binds at HSP90’s C-terminal nucleotide-binding pocket, suggesting that WASF3 is likely not a client protein for HSP90.
Treatment of PC3 cells with 17-AAG for 24-h did, however, lead to markedly decreased WASF3 phosphoactivation and ABL levels, even in the presence of PDGF. To investigate the interaction between HSP90 and ABL, IP, and Western blot analysis of PC3 cells was performed using anti-ABL antibody and confirmed the presence of HSP90 in the complex. Importantly, when 17-AAG was used to inhibit HSP90, ABL levels were undetectable. Taken together, these results indicate that ABL is a client protein of HSP90 and that through HSP90 targeting, WASF3 phosphoactivation is inhibited as a result of ABL kinase destabilization, leading to a decrease in cell invasion and metastasis potential [24].
2.3.5. Regulation by Human Epidermal Growth Factor Receptor 2/3 (HER2/HER3) Signaling Axis
HER2 (ERBB2/neu) and HER3 (ERBB3) are receptor tyrosine kinases implicated in cancer invasion and metastasis. Of these two, HER2 is overexpressed in 20–30% of breast cancers and is strongly associated with poor patient prognosis [31]. While HER3 has also been linked to worse cancer patient outcomes, it contrasts with its family members by lacking tyrosine kinase activity [32]. Through heterodimerization with, for example, HER2, however, HER3 can contribute to signaling activation [32]. In fact, our studies have shown that WASF3-mediated cancer invasion is contingent upon the HER2/HER3 heterodimer, which facilitates WASF3 phosphoactivation and transcription [33]. Specifically, the functional activation of HER2 using NRG was seen to induce the phosphorylation of WASF3 in HER2-positive SKBR3 breast cancer cells. Through confocal microscopy and IP, visualization of WASF3 recruitment to the membrane and interaction with HER2, as well as HER2’s presence in the WASF3 immunocomplex, was also achieved.
After investigating the expression profile and phosphorylation status of HER2 and HER3, it was found that, unlike HER2, HER3’s activation and presence in the WASF3 immunocomplex were contingent upon NRG treatment. Moreover, using siRNA knockdown, it was demonstrated that WASF3 phosphorylation required the expression of both HER2 and HER3, which suggests a key role for HER2/HER3 heterodimerization in WASF3 activation. Blocking HER2 binding using Herceptin also significantly inhibited WASF3 phosphorylation and hindered NRG-induced invasion in WASF3 overexpressing cells. Importantly, this observation was not seen with the treatment of Erlotinib, allowing the influence of EGFR to be excluded. Furthermore, unlike the long-term treatment of NRG which resulted in a four-fold increase in WASF3 protein levels, EGFP treatment did not affect WASF3 levels. These findings suggested that NRG and HER2 are involved in the transcriptional regulation of WASF3, which RT-PCR later confirmed, showing the mRNA levels of both WASF3 and WASF1 to be increased after long-term exposure to NRG [33].
In previous studies, we have also identified STAT3 to be a key regulator of WASF3 expression. Again using SKBR3 cells, we found that NRG stimulation significantly increases STAT3 phosphorylation and that blocking the activation of either STAT3 or JAK2 prevents NRG-induction of WASF3, indicating that WASF3 activation is dependent on HER2/HER3-JAK2/STAT3 signaling [33]. Finally, invasion and metastasis were also evaluated in vitro and in vivo using MCF7 cells and mice and were found to be significantly enhanced following the co-expression of WASF3 and HER2, further demonstrating the crucial role of HER2-WASF3 in cancer metastasis [33].
References
- Guan, X. Cancer metastases: Challenges and opportunities. Acta pharmaceutica sinica B 2015, 5, 402–418.
- Kurisu, S.; Takenawa, T. The WASP and WAVE family proteins. Genome Biol 2009, 10, 226.
- Schaks, M.; Singh, S.P.; Kage, F.; Thomason, P.; Klünemann, T.; Steffen, A.; Blankenfeldt, W.; Stradal, T.E.; Insall, R.H.; Rottner, K. Distinct Interaction Sites of Rac GTPase with WAVE Regulatory Complex Have Non-redundant Functions in Vivo. Curr Biol 2018, 28, 3674–3684.e6.
- Bompard, G.; Caron, E. Regulation of WASP/WAVE proteins: Making a long story short. J Cell Biol 2004, 166, 957–962.
- Frugtniet, B.; Jiang, W.G.; Martin, T.A. Role of the WASP and WAVE family proteins in breast cancer invasion and me-tastasis. Breast Cancer (Dove Med Press) 2015, 7, 99–109.
- Smith, B.A.; Daugherty-Clarke, K.; Goode, B.L.; Gelles, J. Pathway of actin filament branch formation by Arp2/3 complex revealed by single-molecule imaging. Proc Natl Acad Sci U S A 2013, 110, 1285–1290.
- Yamaguchi, H.; Lorenz, M.; Kempiak, S.; Sarmiento, C.; Coniglio, S.; Symons, M.; Segall, J.; Eddy, R.; Miki, H.; Takena-wa, T. et al. Molecular mechanisms of invadopodium formation: The role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol 2005, 168, 441–452.
- Kurisu, S.; Suetsugu, S.; Yamazaki, D.; Yamaguchi, H.; Takenawa, T. Rac-WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene 2005, 24, 1309–1319.
- Takahashi, K.; Suzuki, K. WAVE2, N-WASP, and Mena facilitate cell invasion via phosphatidylinositol 3-kinase-dependent local accumulation of actin filaments. J Cell Biochem 2011, 112, 3421–3429.
- Taniuchi, K.; Furihata, M.; Naganuma, S.; Saibara, T. WAVE2 is associated with poor prognosis in pancreatic cancers and promotes cell motility and invasiveness via binding to ACTN4. Cancer Med 2018, 7, 5733–5751.
- Sossey-Alaoui, K.; Li, X.; Cowell, J.K. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. Journal of Biological Chemistry 2007, 282, 26257–26265.
- Wu, J.; Wang, G.C.; Chen, X.J.; Xue, Z.R. Expression of WASF3 in patients with non-small cell lung cancer: Correlation with clinicopathological features and prognosis. Oncology letters 2014, 8, 1169–1174.
- Huang, S.; Huang, C.; Chen, W.; Liu, Y.; Yin, X.; Lai, J.; Liang, L.; Wang, Q.; Wang, A.; Zheng, C. WAVE3 promotes pro-liferation, migration and invasion via the AKT pathway in pancreatic cancer. Int J Oncol 2018, 53, 672–684.
- Li, X.; Geng, J.; Ren, Z.; Xiong, C.; Li, Y.; Liu, H. WAVE3 upregulation in esophageal squamous cell carcinoma and its ef-fect on the migration of human esophageal cancer cell lines in vitro. Molecular Medicine Reports 2020, 22, 465–473.
- Teng, Y.; Mei, Y.; Hawthorn, L.; Cowell, J.K. WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene 2014, 33, 203–211.
- Wang, W.; Kansakar, U.; Markovic, V.; Wang, B.; Sossey-Alaoui, K. WAVE3 phosphorylation regulates the interplay be-tween PI3K, TGF-β, and EGF signaling pathways in breast cancer. Oncogenesis 2020, 9, 1–13.
- Nie, Y.; Hu, S.; Liu, S.; Fang, N.; Guo, F.; Yang, L.; Liang, X. WASF3 expression correlates with poor prognosis in gastric cancer patients. Future oncology 2019, 15, 1605–1615.
- Ji, Y.; Li, B.; Zhu, Z.; Guo, X.; He, W.; Fan, Z.; Zhang, W. Overexpression of WAVE3 promotes tumor invasiveness and confers an unfavorable prognosis in human hepatocellular carcinoma. Biomedicine & Pharmacotherapy 2015, 69, 409–415.
- Bledzka, K.; Schiemann, B.; Schiemann, W.P.; Fox, P.; Plow, E.F.; Sossey-Alaoui, K. The WAVE3-YB1 interaction regu-lates cancer stem cells activity in breast cancer. Oncotarget 2017, 8, 104072.
- Park, G.B.; Kim, D. TLR5/7-mediated PI3K activation triggers epithelial-mesenchymal transition of ovarian cancer cells through WAVE3-dependent mesothelin or OCT4/SOX2 expression. Oncol Rep 2017, 38, 3167–3176.
- Sossey-Alaoui, K.; Ranalli, T.A.; Li, X.; Bakin, A.V.; Cowell, J.K. WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3, and MMP-9 expression. Exp Cell Res 2005, 308, 135–145.
- Teng, Y.; Ghoshal, P.; Ngoka, L.; Mei, Y.; Cowell, J.K. Critical role of the WASF3 gene in JAK2/STAT3 regulation of can-cer cell motility. Carcinogenesis 2013, 34, 1994–1999.
- Ghoshal, P.; Teng, Y.; Lesoon, L.A.; Cowell, J.K. HIF1A induces expression of the WASF3 metastasis-associated gene un-der hypoxic conditions. International journal of cancer 2012, 131, E905–E915.
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activa-tion of WASF3 metastasis-promoting protein. Journal of Biological Chemistry 2012, 287, 10051–10059.
- Jin, H.; Xie, Q.; Guo, X.; Xu, J.; Wang, A.; Li, J.; Zhu, J.; Wu, X.-R.; Huang, H.; Huang, C. p63α protein up-regulates heat shock protein 70 expression via E2F1 transcription factor 1, promoting Wasf3/Wave3/MMP9 signaling and bladder can-cer invasion. Journal of Biological Chemistry 2017, 292, 15952–15963.
- Baudier, J. ATAD3 proteins: Brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol Rev Camb Philos Soc 2018, 93, 827–844.
- Lang, L.; Loveless, R.; Teng, Y. Emerging Links between Control of Mitochondrial Protein ATAD3A and Cancer. Interna-tional journal of molecular sciences 2020, 21, 7917.
- Teng, Y.; Ren, X.; Li, H.; Shull, A.; Kim, J.; Cowell, J.K. Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene 2016, 35, 333–343.
- Sossey-Alaoui, K.; Li, X.; Ranalli, T.A.; Cowell, J.K. WAVE3-mediated cell migration and lamellipodia formation are reg-ulated downstream of phosphatidylinositol 3-kinase. J Biol Chem 2005, 280, 21748–21755.
- Taylor, M.A.; Davuluri, G.; Parvani, J.G.; Schiemann, B.J.; Wendt, M.K.; Plow, E.F.; Schiemann, W.P.; Sossey-Alaoui, K. Upregulated WAVE3 expression is essential for TGF-β-mediated EMT and metastasis of triple-negative breast cancer cells. Breast cancer research and treatment 2013, 142, 341–353.
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new ad-vances in therapy. Chemotherapy research and practice 2012, 2012.
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta pharmaceutica sinica B 2018, 8, 503–510.
- Teng, Y.; Pi, W.; Wang, Y.; Cowell, J.K. WASF3 provides the conduit to facilitate invasion and metastasis in breast cancer cells through HER2/HER3 signaling. Oncogene 2016, 35, 4633–4640.

