 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Vittoria Dieci | + 3685 word(s) | 3685 | 2021-01-26 05:05:04 | | | |
| 2 | Vivi Li | -2 word(s) | 3683 | 2021-02-02 08:48:14 | | |
Video Upload Options
In recent decades, the increasing interest in the field of immunotherapy has fostered an intense investigation of the breast cancer (BC) immune microenvironment. In this context, tumor-infiltrating lymphocytes (TILs) have emerged as a clinically relevant and highly reproducible biomarker capable of affecting BC prognosis and response to treatment. Indeed, the evaluation of TILs on primary tumors proved to be strongly prognostic in triple-negative (TN) BC patients treated with either adjuvant or neoadjuvant chemotherapy, as well as in early TNBC patients not receiving any systemic treatment, thus gaining level-1b evidence in this setting. In addition, a strong relationship between TILs and pathologic complete response after neoadjuvant chemotherapy has been reported in all BC subtypes and the prognostic role of higher TILs in early HER2-positive breast cancer patients has also been demonstrated. The interest in BC immune infiltrates has been further fueled by the introduction of the first immune checkpoint inhibitors in the treatment armamentarium of advanced TNBC in patients with PD-L1-positive status by FDA-approved assays. However, despite these advances, a biomarker capable of reliably and exhaustively predicting immunotherapy benefit in BC is still lacking, highlighting the imperative need to further deepen this issue. Finally, more comprehensive evaluation of immune infiltrates integrating both the quantity and quality of tumor-infiltrating immune cells and incorporation of TILs in composite scores encompassing other clinically or biologically relevant biomarkers, as well as the adoption of software-based and/or machine learning platforms for a more comprehensive characterization of BC immune infiltrates, are emerging as promising strategies potentially capable of optimizing patient selection and stratification in the research field.
1. Introduction
Breast cancer (BC) is not considered a highly immunogenic tumor type, especially if compared with melanoma or lung cancer. However, in recent decades, it has been consistently reported that the BC tumor microenvironment (BC-TME) encompasses a wide range of cell populations of both the innate and adaptive immune systems, which have been reported to be biologically/clinically relevant to varying degrees [1], as summarized in Figure 1. Immune infiltrates in BC encompass immune cells both directly in contact with tumor cells, known as “intratumoral” (int), and within the surrounding stroma, known as “stromal” (str). The evaluation of BC immune infiltrates relies on, among other things, morphological evaluation of immune cells on H&E-stained tumor samples, immunohistochemical staining for specific subsets of immune cells evaluated by classic semiquantitative scoring or by digital pathology, multiplexed fluorescent immunohistochemistry with multispectral imaging that simultaneously identifies and quantifies multiple immune cell subsets in a single formalin-fixed paraffin-embedded (FFPE) slide and provides information on the distribution of immune infiltrates, flow cytometry on fresh tissue and computational tools for immune cell quantification from transcriptomics data.
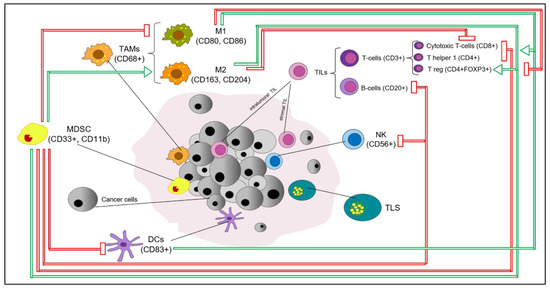
Figure 1. Key immune cell subsets in breast cancer tumor microenvironment. The production of IFNγ by CD4+ Th1 cells mediates the expansion, differentiation and activation of CD8+ tumor-infiltrating lymphocytes (TILs), which subsequently release cytotoxic cytokines and directly kill cancer cells (via recognition of specific tumor-associated antigens on the surface of antigen presentation cells (APCs) or cancer cells); CD4+FOXP3+ TILs represent immunosuppressive mediators through the inhibition of CD8+ T cells, CD4+ Th1 cells, APCs and natural killer cells (NKs); M1 tumor-associated macrophages (M1-TAMs) are associated with Th1 cytotoxic immune response, thus exhibiting antitumor properties; M2-TAMs contribute to the activation of Th2 immune response, thus showing an immunosuppressive role (e.g., suppression of T cell function); NKs are cytotoxic members of the innate immune system (release of cytotoxic cytokines and direct killing of cancer cells); dendritic cells (DCs) are antigen-presenting cells and are crucial players of the adaptive immune system; myeloid-derived suppressor cells (MDSCs) represent immature myeloid cells (possibly originated from bone marrow precursors) with an immunosuppressive function via the inhibition of T cells, B cells, NKs, M1-TAMs and DCs. The recruitment and accumulation of immunosuppressive mediators into the tumor bed is mediated by the secretion of cytokines and chemokines (e.g., IL6, IL1-β, TGF-1β, CCL2) by tumor cells. Red and green arrows reflect inhibitory and stimulatory relationships, respectively. Abbreviations: IFNγ, interferon gamma; TILs, tumor-infiltrating lymphocytes, APCs, antigen presentation cells, NKs, natural killer cells, DCs, dendritic cells, TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells; TLS, tertiary lymphoid structure.
BC immunogenicity is highly heterogeneous, with different BC subtypes showing different degrees of immune infiltration. In detail, the most biologically aggressive subtypes, namely, triple-negative BC (TNBC) and HER2-positive (HER2+) BC, are characterized by high genomic instability and tumor mutational burden (TMB), both fueling the generation of neoantigens, ultimately fostering the antitumor immune activity. However, an inverse association between TMB/genomic heterogeneity and levels of immune infiltrates in TNBC subtypes has recently been suggested, with immune-rich tumors showing lower degrees of clonal genomic heterogeneity, lower neoantigen loads and somatic mutations and fewer somatic copy number alterations [2]. While seemingly counterintuitive, this observation may reflect the elimination of immunogenic clones by an effective antitumor immune surveillance, thus resulting in lower clonal genomic heterogeneity. Conversely, higher clonal heterogeneity may reflect the escape phase of the immunoediting process, where the selection of cancer clones results in reduced immunogenicity.
Both cytotoxic treatments and anti-HER2 agents are known to be capable of further activating the immune system through immunogenic cell death and antibody-dependent cellular cytotoxicity (ADCC), respectively. In addition, in HER2+ BC, oncogene addiction may trigger the immune system, with HER2 itself acting as a tumor-associated neoantigen [3]. Hormone receptor-positive (HR+)/HER2-negative (HER2−) BC, also known as luminal-like BC, is traditionally considered to be less immunogenic than TNBC and HER2+ BC, given the lower genomic instability and mutational load [3][4][5]. However, available evidence suggests that the immunogenicity of this BC subtype may rely on subtler mechanisms reflecting the complex and dynamic relationship between HR+ BC cells, inflammatory mediators, estrogen levels, endocrine treatments and menopausal status [6].
2. Adaptive Immunity
2.1. Tumor infiltrating lymphocytes
In recent decades, the morphological evaluation of immune infiltrates in BC has gained tremendous interest in the light of accumulating high-quality evidence supporting TIL clinical validity in BC. The prevalence of TILs is heterogeneous across different BC subtypes, with TNBC and HER2+ BC typically exhibiting greater TIL infiltration as compared to the luminallike BC subtype [7].
An additional source of heterogeneity is represented by the disease setting. Indeed, it has been suggested that TIL infiltration tends to weaken throughout the natural history of BC from the early to advanced stages. In particular, it has been consistently reported that
overall TIL levels are not only lower in patients with advanced disease as compared to the early setting, but also in heavily treated advanced BC patients as compared to those treated in the first-line setting for their metastatic disease [8][9][10][11]. Moreover, heterogeneity in TIL levels has also been observed within different sites of BC metastases, with the lungs showing the highest degree of TIL infiltration, while the liver and skin show the lowest [10][12][13].
The ever-growing interest towards the evaluation of TILs fostered the development of the International Working Group on Immuno-Oncology Biomarkers, aiming at providing a standardized methodology for TIL assessment in BC samples, in order to improve consistency and reproducibility across studies, in preparation for TIL clinical implementation, also given the endorsement of TIL quantification and reporting in TNBC and HER2+ BC by the St Gallen Consensus Conference (TNBC), WHO (both TNBC and HERBC2+) and ESMO 2019 Guidelines [14][15]. The clinical validity of TILs as a prognostic marker and potential clinical utility in patients with early breast cancer is summarized in Table IV
Table IV. Summary of clinical validity of TILs as a prognostic marker and potential clinical utility in patients with early breast cancer
|
BC Subtype |
Clinical validity (prognostic) |
LoE |
Evaluation endorsed by guidelines |
Potential clinical utilityb (to be demonstrated) |
|
TNBC |
High TILs are associated with improved outcome [16][17][18][19][20][21][22][23][24]
High TILs are associated with increased pCR rate after neoadjuvant chemotherapy[25]
In TNBC, high TILs on residual disease after neoadjuvant chemotherapy are associated with improved outcome[26][27] |
IBa |
Yes: Expert Opinion at the 16th St Gallen International Breast Cancer Conference[15]; ESMO 2019 Early Breast Cancer guidelines[16]; WHO classification of Tumors, Breast Tumors, 5th edition; |
Integration with other clinicopathological variables to guide treatment de-escalation in low-risk patients (i.e. no anthracyclines or even no treatment; prognostic tool available at www.tilsinbreastcancer.org).
Risk stratification in post-neoadjuvant setting based on TILs and RCB to guide the decision of further adjuvant treatment.
Stratification factor in clinical trials. |
|
HER2+ |
Yes: ESMO 2019 Early Breast Cancer guidelines[16]; WHO classification of Tumors, Breast Tumors, 5th edition. |
Integration in multiparametric scores to guide treatment escalation and de-escalation (i.e. HER2DX).
Stratification factor in clinical trials. |
||
|
HR+/HER2- |
Not demonstrated: conflicting results from studies in the adjuvant setting; high TILs associated with increased pCR rate after neoadjuvant chemotherapy, but less favorable survival[28] |
- |
No |
Unknown |
The interest in BC immune infiltrates has been further fueled by the introduction of the first immune checkpoint inhibitors in the treatment armamentarium of advanced TNBC in patients with PD-L1-positive status by FDA-approved assays. Indeed, at present, the only established predictive biomarker for immunotherapy efficacy in metastatic BC is represented by PD-L1 expression, as emerged in the context of randomized phase III trials for TNBC BC. Beyond PD-L1 expression, several other biomarkers proved to be potentially capable of improving our ability to select patients for immunotherapy. Focusing on BC immune infiltrates, interestingly, accumulating evidence suggests that the morphologic evaluation of TILs may have a role in this regard. Notably, it has been consistently reported that PD-L1 expression is predominant on immune cells rather than tumor cells[12][29]. In addition, it has been reported that PD-L1 expression and TIL levels are strongly correlated with each other, especially in the TNBC subtype, thus suggesting that the simple morphological evaluation of TILs may serve as a surrogate reflecting a general state of immune activation[12][30]. Table II summarizes results from translational analyses of several trials testing diverse immune checkpoint blockade strategies, which suggested a potentially clinically relevant role of TILs in BC patients treated with immunotherapy either in the early or advanced setting.
Table II. Summary of phase II/III clinical trials investigating immunotherapy in BC with results from TIL analysis available
|
Trial (design) |
Setting, BC subtype |
Treatment arms |
TIL variable (cutoffa) |
Outcomes |
Resultsc |
|
Keynote-086[31] (II)e |
Advanced, TN |
Pembrolizumab (2 cohorts) |
Binary (median) |
ORR DCR |
Combined cohort: OR for ORR=1.26 (CI 1.03-1.55), p=0.01; OR for CDR 1.22 (CI 1.02-1.46), p= 0.01 |
|
Impassion-130[29] (III) |
Advanced, TN |
NabP-Atezo NabP-Pbo |
Binary (10%) |
PFS OS |
Predictive for Atezo benefit only in PD-L1+: PD-L1+/strTILs lowà PFS: HR 0.74 (0.54-1.03), p=0.07, OS: HR 0.65 (0.41-1.02) p=0.06. PD-L1+/strTILs high à PFS: HR 0.53 (0.38-0.74), p<0.005; OS: HR 0.57 (0.35-0.92), p=0.02. PD-L1-/strTILs high à PFS: HR 0.99 (0.62-1.57), p=0.97; OS: HR 1.53 (0.76-3.08), p=0.24 |
|
Keynote-119 (III)[32] |
Advanced, TN |
Pembro CT PFCb |
Continuous, Binary (5%) |
BOR DCR PFS OS |
Positive association with clinical outcomes only in Pembro arm (p<0.05) TILs <5% à Pembro vs CT: median OS 5.9 vs 8.8 months, HR 1.50 (1.14-1.97) TILs ≥5% à Pembro vs CT: median OS 12.5 vs 11.3 months, HR 0.75 (0.59-0.96) |
|
Panacea[10] (Ib-II) |
Advanced, HER2+ |
Trastuzumab-Pembro |
Continuous |
OR DCR |
Positive association with ORR (p=0.006) and DCR (p=0.0006) |
|
KATE-2 |
Advanced, HER2+ |
TDM1-Atezo TDM1-Pbo |
Binary (5%) |
OS |
Borderline positive association with OS in the Atezolizumab arm TILs<5% à Atezo vs Placebo: 1-year OS 84.2% vs 100%, HR 1.43 (0.51-4.01) TILs≥5% à Atezo vs Placebo: 1-year OS 90.8% vs 83.5%, HR 0.55 (0.26-1-12) |
|
GeparNuevo (II R)[35] |
Neoadjuvant, TN |
Durvae à Durva-NabPà Durva-EC Pboà Pbo-NabPà Pbo-EC |
Continuous Categorical (10%, and 60%) |
pCR |
strTILs (continuous): positive association with pCR, no predictive for Durva benefit OR 1.23 (1.04-1.6), p=0.019 in Durva arm; OR=1.39 (1.12-1.74), p=0.003 in Placebo arm. intTILs: increase between baseline and end of the window phase associated with Durva benefit. OR 9.36 (1.26-69.65) p=0.029 |
|
NeoTRIPaPDL1 (III)[36] |
Neoadjuvant, TN |
Cb-NabP-Atezo Cb-NabP |
Binary (40%) |
pCR |
Positive association with pCR, no predictive role Atezo arm: strTILs ≥40% vs <40% à pCR 71.43% vs 28.07%, p=0.001 CT arm: strTILs ≥40% vs <40% à pCR 63.16% vs 33.9%, p=0.009 No interaction with treatment arm. |
|
GIADA (II)[37] |
Neoadjuvant, Luminal-Bf |
EC à Nivo-ET |
Continuous |
pCR |
Positive association with pCR (p<0.001) |
2.1. Tumor infiltrating Dendritic Cells (DCs)
DCs represent antigen-presenting cells and are crucial players of the adaptive immune system. Interestingly, an impairment of their function, mostly in terms of defective antigen presentation, has been observed in patients with cancer[38]. It has been suggested that
DCs (S100+) in the TME show a compartmentalization pattern, with immature DCs (CD1a-) generally residing within the tumor and mature DCs (CD83+) typically adhering selectively to the peritumoral area[39]. In addition, the presence of DCs was reported to well correlate
with other immune biomarkers such as general TILs and T-regulatory and T-cytotoxic subsets, as well as tertiary lymphoid structures[40].
Despite their well-recognized biological relevance, their role in BC is largely unexplored. The presence of S100+ tumor-infiltrating DCs has been correlated to unfavorable clinicopathologic features, such as higher tumor grade, larger tumor size, nodal involvement and HR-negative status[41][42], while an inverse association between DCs and negative nodal status has been suggested when specifically distinguishing mature DCs (CD83+)[43]. Preliminary data also suggest a prognostic role of tumor-infiltrating DCs in BC patients.
In detail, a retrospective analysis of 130 tumor samples from unselected early BC patients undergoing curative surgery showed a significant and independent positive relationship between higher mature DCs and longer RFS and OS[43]. Interestingly, the mature DC prognostic value was reported to be even larger when considering BC patients with nodal involvement. This observation was further confirmed in a large retrospective cohort of 681 TNBC patients, where the presence of DCs on pre-therapeutic tumor samples was found to be prognostic (in terms of RFS and OS) only in node-positive patients[41]. These data overall highlight a promising prognostic role of DCs in early BC patients. In particular, although preliminary, available evidence suggests that the presence of DCs may help to identify a subgroup of patients with good prognosis despite the presence of nodal involvement at baseline. For this reason, further research in this field is highly encouraged.
3. Innate Immunity
3.1. Tumor-Associated Macrophages (TAMs)
Although TAMs represent a highly heterogeneous immune cell population, they can be commonly dichotomized in two polarized phenotypes, M1 and M2, with the former traditionally associated with antitumor effects, and the latter typically showing protumorigenic characteristics. Surface markers of M1-polarized TAMs are, among others, the costimulatory molecules CD80 and CD86, while the expression on the TAM surface of CD163, CD204 and CD206 is typically associated with M2 polarization. CD68 is instead considered a pan-macrophage marker.
In recent decades, TAMs have gained increasing attention in the light of accumulating preclinical evidence suggesting their involvement in BC tumorigenesis, metastasis and resistance to treatments. In detail, an association between TAMs and protumorigenic processes has been reported, including the activation of oncogenic pathways, the induction of epithelial-to-mesenchymal transition and the promotion of tumor-associated neovascularization [44]. In addition, results from several studies on preclinical BC models suggest that TAMs recruitment into the BC-TME as well as their polarization into an M2 phenotype could hamper the activity of cytotoxic therapies, endocrine treatments, anti-HER2-targeted agents and even immune checkpoint blockade. Although preclinical evidence overall highlights that TAMs represent a biologically relevant player in the context of the BC-TME, results from clinical studies, deepening their role in BC patients, are scattered and broadly inconsistent.
The infiltration of TAMs in the BC-TME has been generally associated with unfavorable clinicopathologic features, such as higher tumor grade, vascular invasion and greater tumor burden (larger tumor size and lymph node involvement), as well as hormone receptor-negative, HER2+ and basal-like phenotypes [45][46][47]. Notably, the evaluation of tumor-associated macrophage activity reflected by the expression of the CD68 gene is shared by several clinically validated genomic assays, such as the 21-gene assay (Oncotype Dx®) and the 70-gene assay (Mammaprint®) [48]. However, when investigating the possible prognostic role of infiltrating TAMs—either when considered as unpolarized TAMs or as selected subpopulations—inconclusive results have been obtained.
In detail, TAMs have been retrospectively reported to predict unfavorable survival rates in terms of DFS, BCSS and OS in unselected BC [47][49], as well as in ER+ or TNBC subtypes [50][51]. Similarly, non-polarized TAMs have been suggested as an independent negative prognostic factor for worse cumulative survival in two large retrospective studies, adopting a computational approach (CIBERSORT) for the analysis of more than 18,000 early BC samples [52][53]. However, an association in the opposite was shown in a large retrospective analysis (n = 478) of early BC patients, where increasing levels of TAMs (CD68+) were positively and independently associated with both DFS and BCSS. In addition, it should be noted that several other studies failed to report any association between unselected TAMs (CD68+) and prognosis [54][55].
As far as M2-polarized (CD163+) TAMs are concerned, results from several retrospective series support their negative prognostic role in unselected early BC patients as well as in TNBC and HER2+ subgroups [47][55][56][57]. Interestingly, the negative association between M2-TAMs and prognosis was further confirmed in a large series of almost 11,000 primary tumor samples, where the authors, by applying a CIBERSORT approach, found an association between M2-TAMs and worse cumulative survival in ER-BC patients [53]. These results appear consistent with the notion that M2 polarization of TAMs may unbalance the BC-TME equilibrium by establishing an immunosuppressive milieu, thus facilitating tumor immune escape.
Nevertheless, a prognostic association in the opposite direction has also been suggested in ER-BC patients, thus further highlighting the need for a more in-depth evaluation of the TAM prognostic value [58].
TAMs’ role in early BC patients has also been deepened regarding the potential association with systemic treatment response.
In detail, both increasing M2- and M1-polarized TAMs have been found to be positively associated with pCR rates in several series of unselected locally advanced BC patients receiving neoadjuvant chemotherapy [52][57]. In contrast, the gene expression profiling of more than 11,000 early BC tumor samples (CIBERSORT approach) suggested that baseline M2-polarized TAMs were capable of predicting the subsequent failure to achieve pCR after neoadjuvant chemotherapy. On the other hand, several other authors failed to report a significant association between baseline TAMs and pCR. In particular, in the translational analysis of 150 baseline tumor samples from a phase II trial of anthracycline–taxane-based treatment (+ bevacizumab), no significant association between M2-TAMs (CD163+) and pCR rates was reported [59].
It is currently unknown whether the inconsistency and inconclusiveness of results on the possible prognostic and/or predictive role of TAMs reflect an actual lack of biological/clinical relevance in BC or are rather the result of heterogeneity in TAM assessment in terms of assays and cutoff points for classifying high TAM infiltration, as well as TME compartments for TAM inclusion. In addition, the majority of the above-mentioned studies adopted CD68 as a pan-macrophage marker. However, it should be noted that a wide range of other immune cell populations, including granulocytes, fibroblasts, lymphocytes and dendritic cells, may also express CD68 on their surface, thus further complicating the interpretation of these results [41].
From a therapeutic point of view, several strategies adopting TAMs as a target have been or are currently being investigated, encompassing inhibition of TAM recruitment, promotion of TAM killing, modulation of TAM polarization and reduction in protumorigenic products released from TAMs. However, although preliminary, available results from pivotal trials testing anti-TAM-targeted strategies overall suggest a limited clinical activity/efficacy in BC patients. Ongoing trials currently investigating therapeutic strategies adopting TAMs or their function as a target are summarized in Table 3.
Table 3. Summary of strategies targeting TAMs which are currently under investigation in breast cancer preclinical and clinical studies.
| Mechanism of Action | Target | Drug | Partners | Trials |
|---|---|---|---|---|
| TAM killing | Membrane cell receptor activation | Trabectidine | CT, PARP inhibitor | NCT00050427 NCT00580112 NCT03127215 |
| CSF1-CSF1R inhibition | Emactuzumab Pexidartinib Lacnotuzumab Cabiralizumab |
CT, immune checkpoint inhibitors | NCT02323191 NCT02760797 NCT01494688 NCT01596751 NCT01525602 NCT01042379 NCT02435680 NCT02807844 NCT03285607 NCT04331067 |
|
| Inhibition of TAM recruitment | CCL2-CCR2 inhibition | Carlutumab | CT | NCT01204996 |
| Modulation of TAM polarization (from M2 into M1 TAM phenotype) | Macrophage | Zoledronic Acid | NA | Approved in both early and metastatic settings |
| CD40 stimulation | Selicrelumab | Other anti-TAM agents | NCT02225002 NCT02157831 NCT02665416 NCT02760797 |
|
| CR3 stimulation | 1,3–1,6 β-glucan | Immunotherapy | NCT02981303 | |
| Induction of cancer cell phagocytosis by TAMs | CD47-SIRPα inhibition | TTI-621, ALX148 Hu5F9-G4 |
Immunotherapy, anti-VEGF agents, anti-HER2 agents | NCT02890368 NCT03013218 NCT02216409 NCT02953782 |
| Regulation of antigen presentation by TAMs | TLR7 stimulation | Imiquimod 852A |
CT, RT | NCT00899574, NCT01421017, NCT00821964, NCT00319748 |
| Vaccination | NA | SV-BR-1-GM H2NVAC |
Immunotherapy | NCT04418219, NCT04144023 |
Abbreviations: TAMs, tumor-associated macrophages; CT, chemotherapy; PARP, poly-ADP polymerase; M2pep, M2peptide; CSF1 (R), colony stimulating factor 1 (receptor); CCL2, chemokine C-C motif ligand 2; CCR2, CCL2 receptor; mAb, monoclonal antibody; Fbln7, fibulin 7; CXCL1, C-X-C Motif Chemokine Ligand 1 Pathway; ZnPPIX, zinc protoporphyrin 9; SIRPα, signal regulatory protein alpha; TLR7, toll-like receptor 7; GM-CSF, granulocyte-macrophage colony stimulating factor.
3.2. Natural Killer (NK) Cells
NK cells are cytotoxic members of the innate immune system, representing a crucial mediator of immunosurveillance and elimination in the cancer immunoediting process. However, their role in affecting BC prognosis and/or response to treatment has been poorly investigated. Preliminary data suggest that increased expression of NK-activating genes was associated with better RFS in a small series of unselected early BC patients [60]. In addition, in a small retrospective cohort of BC patients with locally advanced disease undergoing neoadjuvant treatment [61], higher levels of NK cells (CD56+) on pre-therapeutic samples were significantly associated with better pCR rates. The involvement of NKs in the ADCC-mediated mechanism of action of anti-HER2 monoclonal antibodies is well recognized. Interestingly, several strategies aiming at enhancing NK cell effector function (via ADCC) by anti-HER2 agents are currently under investigation and results are awaited [62].
Although promising, available evidence is immature and further efforts should be made in order to deepen the role of NKs in BC biology and clinical behavior.
4. Tertiary Lymphoid Structures (TLS)
TLS represent lymph node-like aggregates (with a germinal well-defined B lymphocyte- and follicular DC-rich area surrounded by a parafollicular T lymphocyte-rich area) which can be found in non-lymphoid organs, including cancer.
So far, data on the clinical relevance of TLS in BC remain speculative. However, in various retrospective series of cancer patients including breast tumors, the presence of TLS—along with general TIL assessment—has been associated with improved prognosis and increased pCR rates after neoadjuvant therapy in unselected BC as well as in patients with HER2+BC or TNBC subtypes receiving standard treatments. In addition, a recent retrospective study evaluating TLS in primary BC samples and paired metastases showed lower TLS in metastatic samples as compared to paired primary tumors, thus further confirming that BC metastases are generally characterized by lower immune infiltration as compared to primary BC [63].
References
- Burugu, S.; Asleh-Aburaya, K.; Nielsen, T.O. Immune infiltrates in the breast cancer microenvironment: Detection, characterization and clinical implication. Breast Cancer 2017, 24, 3–15.
- Karn, T.; Jiang, T.; Hatzis, C.; Sanger, N.; El-Balat, A.; Rody, A.; Holtrich, U.; Becker, S.; Bianchini, G.; Pusztai, L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1707–1711.
- Savas, P.; Caramia, F.; Teo, Z.L.; Loi, S. Oncogene addiction and immunity: Clinical implications of tumour infiltrating lymphocytes in breast cancers overexpressing the HER2/neu oncogene. Curr. Opin. Oncol. 2014, 26, 562–567.
- Bianchini, G.; Gianni, L. The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol. 2014, 15, e58–e68.
- Luen, S.; Virassamy, B.; Savas, P.; Salgado, R.; Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast 2016, 29, 241–250.
- Dieci, M.V.; Griguolo, G.; Miglietta, F.; Guarneri, V. The immune system and hormone-receptor positive breast cancer: Is it really a dead end? Cancer Treat. Rev. 2016, 46, 9–19.
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360.
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404.
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.; De Laurentis, M.; Nanda, R.; Winer, E.; Mukai, H.; Tamura, K.; Armtrong, A.; et al. Phase 2 study of pembrolizumab as first-line therapy for PD-L1-positive metastatic triple-neative breast cancer: Preliminary data from Keynote-086 cohort B. Presented at the ASCO 2017, Chicago, IL, USA, 2–6 June 2017.
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382.
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993.
- Dieci, M.V.; Tsvetkova, V.; Orvieto, E.; Piacentini, F.; Ficarra, G.; Griguolo, G.; Miglietta, F.; Giarratano, T.; Omarini, C.; Bonaguro, S.; et al. Immune characterization of breast cancer metastases: Prognostic implications. Breast Cancer Res. 2018, 20, 62.
- Luen, S.J.; Salgado, R.; Fox, S.; Savas, P.; Eng-Wong, J.; Clark, E.; Kiermaier, A.; Swain, S.M.; Baselga, J.; Michiels, S.; et al. Tumour-infiltrating lymphocytes in advanced HER2-positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: A retrospective analysis of the CLEOPATRA study. Lancet Oncol. 2017, 18, 52–62.
- Burstein, H.J.; Curigliano, G.; Loibl, S.; Dubsky, P.; Gnant, M.; Poortmans, P.; Colleoni, M.; Denkert, C.; Piccart-Gebhart, M.; Regan, M.; et al. Estimating the benefits of therapy for early-stage breast cancer: The St. Gallen International Consensus Guidelines for the primary therapy of early breast cancer 2019. Ann. Oncol. 2019, 30, 1541–1557.
- Loi, S. The ESMO clinical practise guidelines for early breast cancer: Diagnosis, treatment and follow-up: On the winding road to personalized medicine. Ann. Oncol. 2019, 30, 1183–1184.
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569.
- Leon-Ferre, R.A.; Polley, M.Y.; Liu, H.; Gilbert, J.A.; Cafourek, V.; Hillman, D.W.; Elkhanany, A.; Akinhanmi, M.; Lilyquist, J.; Thomas, A.; et al. Impact of histopathology, tumor-infiltrating lymphocytes, and adjuvant chemotherapy on prognosis of triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 167, 89–99.
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann. Oncol. 2019, 30, 1941–1949.
- de Jong, V.; Wang, Y.; Opdam, M.; ter Hoeve, N.; Jozwiak, K.; Hauptmann, M.; Stathonikos, N.; Horlings, H.; Broeks, A.; Michiels, S.; et al. Prognostic value of tumor infiltrating lymphocytes in young triple-negative breast cancer patients who did not receive adjuvant systemic treatment. Presented at ESMO, 2020 Virtual Meeting, Berlin, Germany, 23–24 May 2020.
- Perez, E.A.; Thompson, E.A.; Ballman, K.V.; Anderson, S.K.; Asmann, Y.W.; Kalari, K.R.; Eckel-Passow, J.E.; Dueck, A.C.; Tenner, K.S.; Jen, J.; et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J. Clin. Oncol. 2015, 33, 701–708.
- Krop, I.; Paulson, J.; Campbell, C.; Kiermaier, A.; Andre, F.; Fumagalli, D.; de Haas, S.; Salgado, R.; Denkert, C.; Loibl, S.; et al. Genomic correlates of response to adjuvant trastuzumab and pertuzumab in HER2 positive breast cancer: Biomarker analysis of the Aphinity trial. J. Clin. Oncol. 2019, 37.
- Dieci, M.V.; Conte, P.; Bisagni, G.; Brandes, A.A.; Frassoldati, A.; Cavanna, L.; Musolino, A.; Giotta, F.; Rimanti, A.; Garrone, O.; et al. Association of tumor-infiltrating lymphocytes with distant disease-free survival in the ShortHER randomized adjuvant trial for patients with early HER2+ breast cancer. Ann. Oncol. 2019, 30, 418–423.
- Conte, P.; Frassoldati, A.; Bisagni, G.; Brandes, A.A.; Donadio, M.; Garrone, O.; Piacentini, F.; Cavanna, L.; Giotta, F.; Aieta, M.; et al. Nine weeks versus 1 year adjuvant trastuzumab in combination with chemotherapy: Final results of the phase III randomized Short-HER studydouble dagger. Ann. Oncol. 2018, 29, 2328–2333.
- Prat, A.; Guarneri, V.; Pare, L.; Griguolo, G.; Pascual, T.; Dieci, M.V.; Chic, N.; Gonzalez-Farre, B.; Frassoldati, A.; Sanfeliu, E.; et al. A multivariable prognostic score to guide systemic therapy in early-stage HER2-positive breast cancer: A retrospective study with an external evaluation. Lancet Oncol. 2020, 21, 1455–1464.
- Fumagalli, D.; Venet, D.; Ignatiadis, M.; Azim, H.A., Jr.; Maetens, M.; Rothe, F.; Salgado, R.; Bradbury, I.; Pusztai, L.; Harbeck, N.; et al. RNA Sequencing to Predict Response to Neoadjuvant Anti-HER2 Therapy: A Secondary Analysis of the NeoALTTO Randomized Clinical Trial. JAMA Oncol. 2017, 3, 227–234.
- Demaria, S.; Volm, M.D.; Shapiro, R.L.; Yee, H.T.; Oratz, R.; Formenti, S.C.; Muggia, F.; Symmans, W.F. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin. Cancer Res. 2001, 7, 3025–3030.
- Prowell, T.M.; Beaver, J.A.; Pazdur, R. Residual Disease after Neoadjuvant Therapy—Developing Drugs for High-Risk Early Breast Cancer. N. Engl. J. Med. 2019, 380, 612–615.
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618.
- Emens, L.; Loi, S.; Rugo, H.; Schneeweiss, A.; Dieras, V.; Iwata, H.; Barrios, C.; Nechaeva, M.; Molinero, L.; Nguyen, A.; et al. IMpassion130: Efficacy in immune biomarker subgroup from the global, randomized, double-blind, placebo-controlled, phase III study of atezolizumab + nab-paclitaxel in patients with treatment-naive, locally advanced or metastatic triple-negative breast cancer. Presented at the 2018 San Antonio Breast Cancer Sympsium, San Antonio, TX, USA, 4–7 December 2018.
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380.
- Loi, S.; Adams, S.; Schmid, P.; Cortes, J.; Cescon, D.; Winer, E.; Toppmeyer, D.; Rugo, H.; De Laurentiis, M.; Nanda, R.; et al. Relationship between tumor infiltrating lymphocyte levels and response to pembrolizumab in metastatic triple-negative breast cancer: Results from Keynote-086 trial. Presented at the European Society of Medical Oncology (ESMO) 2017 Congress, Madrid, Spain, 8–12 September 2017.
- Loi, L.; Winer, E.; Lipatov, O.; Im, S.; Goncalves, A.; Cortes, J.; Lee, K.; Schmid, P.; Testa, L.; Witzel, I.; et al. Relationship between Tumor-Infiltrating Lymphocytes (TILs) and Outcomes in the KEYNOTE-119 Study of Pembrolizumab vs Chemotherapy for Previously Treated Metastatic Triple-Negative Breast Cancer (mTNBC); American Association for Cancer Research: Philadelphia, PA, USA, 2020.
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.B.; Im, S.A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol 2020, 21, 1283–1295.
- Emens, L.; Esteva, F.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.; Im, S.; Wang, Y.; Mani, A.; Shah, J.; et al. Overall Survival (OS) in KATE2, a phase 2 study of programmed death ligand 1 (PD-L1) inhibitor Atezolizumab (Atezo) + Trastuzumab Emtansine (T-DM1) vs placebo (PBO) in previously treated HER2+ advanced breast cancer. Ann. Oncol. 2019, 30 (Suppl. 5), v104–v142.
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288.
- Bianchini, G.; Huang, C.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Russo, S.; Ciruelos, E.; et al. Tumour infiltrating lymphocytes (TILs), PD-L1 expression and their dynamics in the NeoTRIPaPDL1 trial. Ann. Oncol. 2020, 31 (Suppl. 4), S1142–S1215.
- Dieci, M.; Guarneri, V.; Bisagni, G.; Tosi, A.; Musolino, A.; Spazzapan, S.; Moretti, G.; Vernaci, G.; Giarratano, T.; Lo Mele, M.; et al. Neoadjuvant chemotherapy and immunotherapy in Luminal B BC: Results of the phase II GIADA trial. Ann. Oncol. 2020, 31, S304–s305.
- Gabrilovich, D.I.; Ishida, T.; Nadaf, S.; Ohm, J.E.; Carbone, D.P. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin. Cancer Res. 1999, 5, 2963–2970.
- Bell, D.; Chomarat, P.; Broyles, D.; Netto, G.; Harb, G.M.; Lebecque, S.; Valladeau, J.; Davoust, J.; Palucka, K.A.; Banchereau, J. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J. Exp. Med. 1999, 190, 1417–1426.
- Lee, H.; Lee, H.J.; Song, I.H.; Bang, W.S.; Heo, S.H.; Gong, G.; Park, I.A. CD11c-Positive Dendritic Cells in Triple-negative Breast Cancer. In Vivo 2018, 32, 1561–1569.
- Lespagnard, L.; Gancberg, D.; Rouas, G.; Leclercq, G.; de Saint-Aubain Somerhausen, N.; Di Leo, A.; Piccart, M.; Verhest, A.; Larsimont, D. Tumor-infiltrating dendritic cells in adenocarcinomas of the breast: A study of 143 neoplasms with a correlation to usual prognostic factors and to clinical outcome. Int. J. Cancer 1999, 84, 309–314.
- Treilleux, I.; Blay, J.Y.; Bendriss-Vermare, N.; Ray-Coquard, I.; Bachelot, T.; Guastalla, J.P.; Bremond, A.; Goddard, S.; Pin, J.J.; Barthelemy-Dubois, C.; et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res. 2004, 10, 7466–7474.
- Iwamoto, M.; Shinohara, H.; Miyamoto, A.; Okuzawa, M.; Mabuchi, H.; Nohara, T.; Gon, G.; Toyoda, M.; Tanigawa, N. Prognostic value of tumor-infiltrating dendritic cells expressing CD83 in human breast carcinomas. Int. J. Cancer 2003, 104, 92–97.
- Lu, J.; Ma, L. The role of tumor-associated macrophages in the development, metastasis and treatment of breast cancer. Pathol. Res. Pract. 2020, 216, 153085.
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955.
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189.
- Tiainen, S.; Tumelius, R.; Rilla, K.; Hamalainen, K.; Tammi, M.; Tammi, R.; Kosma, V.M.; Oikari, S.; Auvinen, P. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology 2015, 66, 873–883.
- Brufsky, A.M.; Davidson, N.E. Multiparametric Genomic Assays for Breast Cancer: Time for the Next Generation? Clin. Cancer Res. 2016, 22, 4963–4965.
- Mahmoud, S.M.; Lee, A.H.; Paish, E.C.; Macmillan, R.D.; Ellis, I.O.; Green, A.R. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J. Clin Pathol. 2012, 65, 159–163.
- Yuan, Z.Y.; Luo, R.Z.; Peng, R.J.; Wang, S.S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Oncol. Targets Ther. 2014, 7, 1475–1480.
- Gwak, J.M.; Jang, M.H.; Kim, D.I.; Seo, A.N.; Park, S.Y. Prognostic value of tumor-associated macrophages according to histologic locations and hormone receptor status in breast cancer. PLoS ONE 2015, 10, e0125728.
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.B.A.G.; van Vugt, M.A.T.M.; de Vries, E.G.E.; Schroder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2016, 109.
- Ali, H.R.; Chlon, L.; Pharoah, P.D.; Markowetz, F.; Caldas, C. Patterns of Immune Infiltration in Breast Cancer and Their Clinical Implications: A Gene-Expression-Based Retrospective Study. PLoS Med. 2016, 13, e1002194.
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020, 111, 47–58.
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101.
- Zhang, Y.; Cheng, S.; Zhang, M.; Zhen, L.; Pang, D.; Zhang, Q.; Li, Z. High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLoS ONE 2013, 8, e76147.
- Kaewkangsadan, V.; Verma, C.; Eremin, J.M.; Cowley, G.; Ilyas, M.; Satthaporn, S.; Eremin, O. The Differential Contribution of the Innate Immune System to a Good Pathological Response in the Breast and Axillary Lymph Nodes Induced by Neoadjuvant Chemotherapy in Women with Large and Locally Advanced Breast Cancers. J. Immunol. Res. 2017, 2017, 1049023.
- Pelekanou, V.; Villarroel-Espindola, F.; Schalper, K.A.; Pusztai, L.; Rimm, D.L. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in ER-positive and -negative cancers. Breast Cancer Res. 2018, 20, 1–10.
- Matikas, A.; Lovrot, J.; Ramberg, A.; Eriksson, M.; Lindsten, T.; Lekberg, T.; Hedenfalk, I.; Loman, N.; Bergh, J.; Hatschek, T.; et al. Dynamic evaluation of the immune infiltrate and immune function genes as predictive markers for neoadjuvant chemotherapy in hormone receptor positive, HER2 negative breast cancer. Oncoimmunology 2018, 7, e1466017.
- Ascierto, M.L.; Idowu, M.O.; Zhao, Y.; Khalak, H.; Payne, K.K.; Wang, X.Y.; Dumur, C.I.; Bedognetti, D.; Tomei, S.; Ascierto, P.A.; et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J. Transl. Med. 2013, 11, 145.
- Verma, C.; Kaewkangsadan, V.; Eremin, J.M.; Cowley, G.P.; Ilyas, M.; El-Sheemy, M.A.; Eremin, O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): Differential restoration of blood profiles by NAC and surgery. J. Transl. Med. 2015, 13, 180.
- Muntasell, A.; Cabo, M.; Servitja, S.; Tusquets, I.; Martinez-Garcia, M.; Rovira, A.; Rojo, F.; Albanell, J.; Lopez-Botet, M. Interplay between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front. Immunol. 2017, 8, 1544.
- Solinas, C.; Boisson, A.; Brown, D.; de Wind, R.; van den Eynden, G.; Garaud, S.; Buisseret, L.; Naveaux, C.; Sotiriou, C.; Larsimont, D.; et al. ESMO 2016 Tumor infiltrating lymphocytes and tertiary lymphoid structures in paired primary tumors and metastases from breast cancer patients. Ann. Oncol. 2016, 27, 545–551.

