 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Izumi Horikawa | + 2758 word(s) | 2758 | 2021-01-14 07:14:59 | | | |
| 2 | Camila Xu | -4 word(s) | 2754 | 2021-01-28 09:30:48 | | | | |
| 3 | Camila Xu | -4 word(s) | 2754 | 2021-01-28 09:31:49 | | | | |
| 4 | Catherine Yang | Meta information modification | 2754 | 2021-09-28 11:16:15 | | |
Video Upload Options
The TP53 gene is a critical tumor suppressor and key determinant of cell fate which regulates numerous cellular functions including DNA repair, cell cycle arrest, cellular senescence, apoptosis, autophagy and metabolism. The delta133p53 isoforms are critical regulators of these biological processes in human physiology and diseases such as cancer.
1. Introduction
TP53 gene is known as “the guardian of the genome” [1] highlighting its crucial role in maintaining genetic stability and preventing cancer formation. p53 is both a central sensor and a master regulator of cellular stresses. Its activation is adapted to the cell type and signal and induces a myriad of cellular responses such as DNA repair, cell cycle arrest, cellular senescence, apoptosis, autophagy and metabolism [2][3]. It is now known that wild-type TP53 encodes, in tissue- and cell type-dependent manners, at least 12 different p53 protein isoforms: p53α (canonical p53), p53β, p53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β and Δ160p53γ [4].
The p53 family members interact with each other and can modulate each other’s expression and biological activities. This includes the p63 and p73 family proteins which can also interact to regulate several common target genes. For example, in the absence of p63 and p73 binding to p53-responsive promoters, p53 fails to induce apoptosis in response to DNA damage demonstrating the significance of p53 family member interactions in p53 pathway activity [5].
2. Regulatory Mechanisms of the Δ133p53 Isoforms
2.1. RNA Level
The mRNAs encoding the Δ133p53 isoforms are produced from the internal TP53 promoter in the intron 4. Each of the Δ133p53 isoforms is translated from the methionine codon 133 and produced through alternative splicing of mRNA from exon-9 to either exon-10 (Δ133p53α), exon-9β (Δ133p53β) or exon-9γ (Δ133p53γ). This results in either the inclusion of the oligomerization and α C-terminal domains (62 amino-acids identical to those of canonical full-length p53α), the β C-terminal domain (10 amino acids), or the γ C-terminal domain (15 amino acids), respectively (Figure 1). In addition, mRNAs encoding the Δ133p53 isoforms can also be translated from the methionine codon 160, which produces each of the Δ160p53 isoforms with the same C-terminal variations.
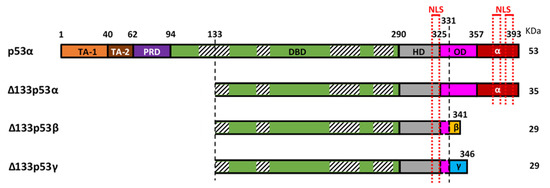
Figure 1. Representation of canonical p53 (p53α) and the three Δ133p53 isoforms (Δ133p53α, Δ133p53β and Δ133p53γ). p53α contains two transactivation domains (TAD1 and TAD2), the proline-rich domain (PRD), the DNA binding domain (DBD), the hinge domain (HD), the oligomerization domain (OD) and the α-domain. Part of the OD and the α-domain can be alternatively spliced to create the β and γ isoforms. The Δ133p53 isoforms lack TA-1, TA-2, PRD and part of the DBD. The DBD contains 4 highly conserved regions (striped boxes) spanning amino-acids 117–142, 171–181, 234–258 and 270–286 respectively, and the Δ133p53 isoforms lack, therefore, the majority of the first conserved region. Three nuclear localization sequences (NLS) are in the C-terminal part (red dotted lines), one in the HD and two in the α-domain, so the β and γ isoforms only have the first one.
The p53 isoforms are expressed in normal human tissues and are modulated by multiple mechanisms. Firstly, epigenetic events regulating the activity of the p53 proximal and internal promoters result in differential expression of Δ133/Δ160 α, β or γ mRNAs in normal human tissues [6]. In addition, the internal TP53 promoter activity is influenced by polymorphisms, including the common pin3 (16-bp insertion in the intron-3) and R72P polymorphisms [7][8][9]. Furthermore, the internal promoter is transactivated by p53α and several p63/p73 family members and is repressed by p68, a co-activator of p53-dependent transcription, particularly following genotoxic stress [8][10][11]. Finally, splicing factors, such as SRSF1 and SRSF3, regulate the alternative splicing of TP53 intron-9 by inhibiting retention of exon-9β/9γ [12][13]. This results in reduced production of the β and γ isoforms. Consistent with this function, treatment with TG003, a small drug inhibitor of the Cdc2-like kinases (Clk), prevents Clk-induced activation of SRSF1 and SRSF3 leading to increased exon-9β/9γ inclusion [13]. In contrast to SRSF1 and SRSF3, SRSF7 has been reported to enhance β splicing in response to ionizing radiation (IR) [14]. In this context, IR reduces SMG1 (a PI3K-like protein) binding on p53 pre-mRNA which enables the recruitment of RPL26. This allows recruitment of SRSF7 which favors the inclusion of exon 9β. Although these splicing factors primarily regulate the production of β/γ isoforms, SRSF1 has also been demonstrated to upregulate Δ133p53α expression without modulating canonical p53α [15], suggesting a possible link between the regulation of alternative transcription and the alternative splicing. However, the exact mechanism of this regulation is not known.
2.2. Protein Level
At the protein level, the differential inclusion of canonical p53 domains in each of the p53 isoforms affects their behavior. The Δ133p53 isoforms lack the first 132 amino-acids and are, therefore, devoid of the two transactivation domains (TADs) and part of the DNA-binding domain (DBD) (Figure 1). The DBD contains four conserved regions, the first of which spans between amino-acids 117 and 142, so the Δ133p53 isoforms lack most of this first region but retain the three others. These four conserved regions coordinate Zn2+ loops structures which are essential for p53 conformation [16][17]. This suggests that the Δ133p53 isoforms may have a different conformation as compared to their full-length counterpart. Although their conformation is not yet known, a recent study of the thermodynamic stability of Δ133p53β demonstrated that it has a higher aggregation propensity but can form a relatively stable complex with p53-specific DNA [18]. This suggests that there may be some differences in Δ133p53β conformation promoting aggregation but not enough to inhibit its binding to p53 response elements. The C-terminal region contains the hinge domain (HD), the oligomerization domain (OD), and the α regulatory domain (Figure 1). In the β and γ isoforms, part of the OD and the α domain are replaced by the β or γ domains, respectively. The lack of the OD in the β and γ isoforms may impair their ability to oligomerize with other isoforms. However, p53β and p53γ can still form DNA-mediated complexes with p53α in the presence of p53-responsive promoter suggesting that the ability of these isoforms to interact with p53α is not completely abolished [13]. In addition, the differences between the α, β or γ domains may impact cellular localization of the isoforms. In p53α, there are three nuclear localization sequences (NLS): one in the HD and two in the α-domain (Figure 1). The Δ133p53α protein, which also contains all three NLS, is primarily located in the nucleus [6]. Although the Δ133p53β lacks two of the three NLS, it contains the HD-associated NLS and is detected in the cytoplasm and in the nucleus where it forms speckles. Interestingly, Δ133p53γ has the same NLS as Δ133p53β; however, it is mostly detected in the cytoplasm. This indicates that β and γ domains are not equivalent and may suggest that either the β peptide modifies the subcellular localization or that the γ peptide counteracts the NLS activity.
Although MDM-2 binds to p53 mainly in the N-terminal TAD, it also contains a secondary binding site in the DBD which is common to all isoforms [19]. Camus et al. confirmed that MDM-2 binds to all p53 isoforms, including the Δ133p53, without promoting their degradation [20]. Furthermore, they reported that all p53 isoforms are ubiquitylated and degraded by the proteasome with different kinetics in a cancer cell line. Hence, Δ133p53γ has the shortest half-life of all the isoforms, about 40 min, while Δ133p53α and Δ133p53β have half-lives of 110 and 135 min, respectively. The half-lives of all three Δ133p53 isoforms were increased upon inhibition of the proteasome by MG-132 treatment, although the increase was smaller for Δ133p53β. It is, therefore, expected that MDM-2 can also block the transcriptional activities of most isoforms either directly or by recruiting corepressors such as hCtBP2 [21][22]. In addition to proteasome degradation, Δ133p53α is primarily degraded by chaperone-mediated autophagy during replicative senescence in normal human cells including fibroblasts, astrocytes and T-cells [23][24][25]. This process may be inhibited by direct interaction of the E3 ubiquitin ligase STUB1/CHIP with Δ133p53α or by pharmacological inhibition of autophagy by bafilomycin A1 [23].
Finally, one of the major regulatory systems of p53 stability and activity is post-translational modifications (PTM). The PTMs occurring on p53α and their implications for its stability and activity have been extensively studied and described [26][27][28]. However, it is not known whether the same modifications occur on Δ133p53 isoforms and, if so, whether they have similar effects. Given that the Δ133p53 isoforms retain several of the PTM sites described in p53α, and since the β (DQTSFQKENC) and γ (MLLDLRWCYFLINSS) C-terminal sequences also contain several serines, threonines, lysines or arginines residues, it is expected that several PTM can occur on all isoforms. In support of this expectation, p53β and p53γ appear as several dots with different isoelectric points when visualized on 2D gels, suggesting different charges due to post-translational modifications [29][30][31][32]. In addition, as mentioned earlier, Camus et al. have reported that all p53 isoforms are ubiquitylated without necessarily promoting their degradation [20]. This suggests that ubiquitylation of p53 isoforms may also be associated with proteasome-independent functions, including the regulation of subcellular location and protein interaction [33][34], further underscoring the potential for PTMs to modulate the stability and activity of p53 isoforms.
3. The Δ133p53 Isoforms in Cancers
Clinical sample studies widely rely on qRT-PCR methods to determine the expression of mRNAs. Therefore, it is important to consider the technical limitations of the current detection methods. The first limitation is the difficulty to detect p53 isoforms expressed at lower abundance in patient samples or in RNA-seq databases [35]. Secondly, to specifically detect and quantify each of Δ133p53α, Δ133p53β and Δ133p53γ mRNAs separately, a simultaneous discrimination of N-terminal events (i.e., the transcriptional initiation for Δ133p53/Δ160p53 versus that for p53α and Δ40p53) and C-terminal events (i.e., α, β or γ splicing) is required. Since Δ133p53α, Δ133p53β and Δ133p53γ mRNAs share a common region spanning 618 base-pairs, such simultaneous discrimination by qRT-PCR would need to amplify long amplicons, which may be difficult to obtain with conventional PCR methods and clinical RNA samples that could be partially degraded. Therefore, earlier clinical studies have only quantitated total expression levels of all three Δ133p53 mRNAs without distinguishing between α, β or γ transcripts (Table 1, upper). Recent technical developments, such as RNAscope (RNA in situ hybridization), chromatography-tandem mass spectrometry and multiplex long amplicon droplet digital PCR, are beginning to circumvent these limitations [36][37][38] (Table 1, lower).
Table 1. Expression of Δ133p53 isoforms in human cancer associated with disease progression, therapy response and patient prognosis.
| Cancer Type | Total Δ133p53 mRNAs (α, β and γ) Association with Disease |
|---|---|
| Colon cancer | Total Δ133p53 mRNAs are downregulated in adenoma vs. normal tissue, but overexpressed in carcinoma vs. adenoma or normal tissue |
| Total Δ133p53 mRNAs expression is correlated with higher risks of recurrence | |
| Elevation of total Δ133p53 mRNAs is associated with shorter disease-free survival | |
| Cholangiocarcinoma | Total Δ133p53 mRNAs upregulation, and decreased TA mRNAs, is associated with shortened overall survival |
| Total Δ133p53 mRNAs reduction sensitizes to 5-Fluorouracil treatment | |
| Lung carcinoma | Total Δ133p53 mRNAs are overexpressed in tumor as compared to adjacent non-cancerous tissue |
| Esophageal Squamous Cell Carcinoma | High tissue or serum total Δ133p53/TAp53 ratio is associated with poor overall and progression-free survival. |
| Ovarian cancer | Total Δ133p53 mRNAs expression in mutant TP53 patients is associated with longer overall survival and disease-free survival |
| Total Δ133p53 mRNAs are associated with increased overall survival (and borderline disease-free survival) independently of TP53 mutation status | |
| Renal cell carcinoma | Wild-type tumors correlate with worse overall survival. Total Δ133p53 mRNAs are downregulated in WT tumors compared to mutant tumors and normal adjacent tissue |
| Cancer Type | Single Specific Δ133p53 Isoform Association with Disease |
| Breast cancer | Δ133p53α mRNA is detected in tumor but not in normal tissue |
| Δ133p53β mRNA detection is associated with worse overall and disease-free survival | |
| Δ133p53β protein is overexpressed in invasive tumor as compared to non-invasive ones | |
| Glioblastoma | Δ133p53β is elevated in glioblastoma malignant cells and is associated with immunosuppressive and chemoresistant environment |
| Prostate cancer | Δ133p53β mRNA is elevated in tumor vs. non-neoplastic tissue and is associated with shorter progression-free survival |
| Melanoma | Increased Δ133p53β mRNA expression is associated with poorer overall survival |
The first part of the table displays the studies in which all three Δ133p53 mRNAs have been quantified as a whole, while the second part of the table displays the studies in which a single specific isoform has been identified.
Given their potency to regulate crucial aspects of the p53 response, investigation of associations between the Δ133p53 isoforms and cancer patient prognosis started shortly after their identification (Table 1). In colorectal cancers, total Δ133p53 mRNAs are downregulated in colon adenomas compared to non-tumor cells, but their expression is upregulated during the progression from adenoma to carcinoma [39]. This suggests that the total Δ133p53 isoforms may not promote benign tumor formation but may play a role in progression from benign to malignant tumors. Consistently, increased expression of total Δ133p53 mRNAs is associated with shorter disease-free survival and correlate with higher risks of recurrence [40][41]. This could be due to Δ133p53α-mediated activation of JAK-STAT3 and RhoA-ROCK signaling promoting colorectal cancer cell growth and invasion [41]. In addition, Δ133p53β protein binds to and inhibits RhoB, thus preventing RhoB-induced apoptosis [40]. This may have important implications in tumor response to therapies such as camptothecin [40]. This is further supported by data from analysis of cholangiocarcinomas in which shortened overall patient survival is correlated with upregulation of total Δ133p53 mRNAs and downregulation of full length p53 mRNAs (p53α/p53β/p53γ) [42]. Interestingly, targeting the Δ133p53 isoforms restores sensitivity to 5-Fluorouracil in resistant cholangiocarcinomas [43]. A possible explanation for this is the ability of Δ133p53α to enhance DNA repair [52][53][54], which counteracts the effect of 5-FU. Critically, Δ133p53α may have a similar impact on other DNA-damage inducing cancer therapies. Similar upregulation of total Δ133p53 mRNAs has been observed in B-cell precursor acute lymphoblastic leukemia [55] and in lung carcinomas as compared to adjacent non-cancerous tissue [44] and is reported to increase cancer cell survival in pathogen-driven cancers such as gastric tumors associated with H. pylori [56]. In the case of gastric cancer, total Δ133p53 mRNAs expression correlates with the NF-κB p65 subunit expression and may promote gastric cell growth [57]. Recently, a high total Δ133p53/TAp53 ratio was shown to be a marker of poor overall and progression-free survival in esophageal squamous cell carcinoma [45]. Furthermore, specific changes in this ratio were correlated to clinical events. For example, the ratio quickly diminished following surgical resection of the tumor and increased upon tumor recurrence. Interestingly, this ratio was measured as circulating RNAs in patient’s serum, which would represent a non-invasive, real-time method to monitor patient’s recurrence.
Importantly, total Δ133p53 mRNAs expression is not always a bad prognostic marker. In mutant TP53 serous ovarian cancer, patients expressing total Δ133p53 mRNAs have longer overall survival and disease-free survival [46][58]. Interestingly, a recent study on high-grade serous ovarian cancer established that high expression of total Δ133p53 mRNAs is correlated with increased overall survival independently of TP53 mutation status [47]. This suggests that the association between total Δ133p53 mRNAs expression and good prognosis in serous ovarian cancers does not depend exclusively on TP53 mutation status. This theory is reinforced by another recent publication which reported that wild-type (WT) TP53 renal cell carcinomas (RCC) have a worse overall survival than those that harbor p53 mutations [48]. Interestingly, total Δ133p53 mRNAs were found downregulated in WT tumors as compared to non-tumor adjacent tissue, while they were unaffected in mutant tumors. This suggests that the worse prognosis in WT RCC may be due to the reduction of WT Δ133p53 expression.
In all the above studies, the clinical associations have been made with the expression level of all Δ133p53 mRNAs. Therefore, these associations cannot be linked to a biological activity of a specific isoform without cell-based functional assays (described below). Nevertheless, some studies using recent techniques of RNA quantification gathered insights into a possible role of individual Δ133p53 isoforms (Table 1, lower). For example, using nested-PCR, Δ133p53α mRNA is detected in breast cancer samples but not in normal breast tissue [6]. However, it has also been shown that Δ133p53α is neither oncogenic nor mutagenic in normal human cells (Table 2) [54], which indicates that it may be involved in cancer progression once developed, but not in cancer formation. In breast cancer patients, detection of Δ133p53β mRNA by nested-PCR is associated with worse overall and disease-free survival [49]. This was explained by its propensity to promote cancer invasion by triggering epithelial to mesenchymal transition [49][50]. Using the new RNAscope in situ hybridization and chromatography-tandem mass spectrometry techniques, increased Δ133p53β expression was found to stimulate glioblastoma and prostate cancer development and aggressiveness by promoting an immunosuppressive and chemo-resistant microenvironment [36][37]. In prostate cancer, increased Δ133p53β mRNA expression is associated with shorter progression-free survival [37]. Its increased expression has also been associated with poorer overall survival in melanoma [51].
Table 2. Δ133p53α is non-mutagenic and non-oncogenic in normal human cells.
| Δ133p53α is | Because Δ133p53α |
|---|---|
| Non-mutagenic | Enhances DNA double-strand break repair |
| Does not induce chromosomal and microsatellite repeats abnormalities | |
| Does not cause increased mutation rate, unlike canonical p53α knockdown | |
| Non-oncogenic | Does not prevent p53α-dependent apoptosis of severely damaged cells |
| Increases replicative lifespan without immortalizing cells | |
| Does not induce malignant transformation |
References
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16, doi:10.1038/358015a0.
- Lane, D.; Levine, A. P53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893, doi:10.1101/cshperspect.a000893.
- Lozano, G.; Levine, A.J. The p53 protein: From cell regulation to cancer; A Cold Spring Harbor Perspectives in Medicine Collection, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 2016.
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039, doi:10.1101/cshperspect.a026039.
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564, doi:10.1038/416560a.
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137, doi:10.1101/gad.1339905.
- Bellini, I.; Pitto, L.; Marini, M.G.; Porcu, L.; Moi, P.; Garritano, S.; Boldrini, L.; Rainaldi, G.; Fontanini, G.; Chiarugi, M.; et al. DeltaN133p53 expression levels in relation to haplotypes of the TP53 internal promoter region. Hum. Mutat. 2010, 31, 456–465, doi:10.1002/humu.21214.
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet de Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.-C. Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826, doi:10.1038/cdd.2011.152.
- Eiholzer, R.A.; Mehta, S.; Kazantseva, M.; Drummond, C.J.; McKinney, C.; Young, K.; Slater, D.; Morten, B.C.; Avery-Kiejda, K.A.; Lasham, A.; et al. Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers 2020, 12, 2472, doi:10.3390/cancers12092472.
- Moore, H.C.; Jordan, L.B.; Bray, S.E.; Baker, L.; Quinlan, P.R.; Purdie, C.A.; Thompson, A.M.; Bourdon, J.-C.; Fuller-Pace, F.V. The RNA helicase p68 modulates expression and function of the Δ133 isoform(s) of p53, and is inversely associated with Δ133p53 expression in breast cancer. Oncogene 2010, 29, 6475–6484, doi:10.1038/onc.2010.381.
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.-C.; Lane, D.P.; Bourdon, J.-C. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258, doi:10.1038/cdd.2010.91.
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.-M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798, doi:10.1038/onc.2012.288.
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.-C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387, doi:10.1038/cdd.2014.73.
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage-induced Alternative Splicing Pathway that Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781, doi:10.1158/2159-8290.CD-16-0908.
- Xie, N.; Chen, M.; Dai, R.; Zhang, Y.; Zhao, H.; Song, Z.; Zhang, L.; Li, Z.; Feng, Y.; Gao, H.; et al. SRSF1 promotes vascular smooth muscle cell proliferation through a Δ133p53/EGR1/KLF5 pathway. Nat. Commun. 2017, 8, 16016, doi:10.1038/ncomms16016.
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564, doi:10.1101/gad.7.12b.2556.
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355, doi:10.1126/science.8023157.
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. 2019, 33, 4225–4235, doi:10.1096/fj.201801973R.
- Ma, J.; Martin, J.D.; Zhang, H.; Auger, K.R.; Ho, T.F.; Kirkpatrick, R.B.; Grooms, M.H.; Johanson, K.O.; Tummino, P.J.; Copeland, R.A.; et al. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry 2006, 45, 9238–9245, doi:10.1021/bi060661u.
- Camus, S.; Ménendez, S.; Fernandes, K.; Kua, N.; Liu, G.; Xirodimas, D.P.; Lane, D.P.; Bourdon, J.-C. The p53 isoforms are differentially modified by Mdm2. Cell Cycle Georget. Tex 2012, 11, 1646–1655, doi:10.4161/cc.20119.
- Thut, C.J.; Goodrich, J.A.; Tjian, R. Repression of p53-mediated transcription by MDM2: A dual mechanism. Genes Dev. 1997, 11, 1974–1986, doi:10.1101/gad.11.15.1974.
- Mirnezami, A.H.; Campbell, S.J.; Darley, M.; Primrose, J.N.; Johnson, P.W.M.; Blaydes, J.P. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr. Biol. CB 2003, 13, 1234–1239, doi:10.1016/s0960-9822(03)00454-8.
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706, doi:10.1038/ncomms5706.
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257, doi:10.1172/JCI70355.
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528, doi:10.1038/cdd.2016.37.
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950, doi:10.1101/cshperspect.a000950.
- DeHart, C.J.; Chahal, J.S.; Flint, S.J.; Perlman, D.H. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell. Proteom. MCP 2014, 13, 1–17, doi:10.1074/mcp.M113.030254.
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346, doi:10.1042/BJ20150517.
- Anensen, N.; Oyan, A.M.; Bourdon, J.-C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3985–3992, doi:10.1158/1078-0432.CCR-05-1970.
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545, doi:10.1038/onc.2011.348.
- Hjelle, S.M.; Sulen, A.; Øye, O.K.; Jørgensen, K.; McCormack, E.; Hollund, B.E.; Gjertsen, B.T. Leukocyte p53 protein biosignature through standard-aligned two-dimensional immunoblotting. J. Proteom. 2012, 76, 69–78, doi:10.1016/j.jprot.2012.07.021.
- Kloster, M.M.; Naderi, E.H.; Haaland, I.; Gjertsen, B.T.; Blomhoff, H.K.; Naderi, S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. Int. J. Oncol. 2013, 42, 1815–1821, doi:10.3892/ijo.2013.1853.
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205, doi:10.1126/science.1127085.
- Schnell, J.D.; Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 2003, 278, 35857–35860, doi:10.1074/jbc.R300018200.
- Mehta, S.; Tsai, P.; Lasham, A.; Campbell, H.; Reddel, R.; Braithwaite, A.; Print, C. A Study of TP53 RNA Splicing Illustrates Pitfalls of RNA-seq Methodology. Cancer Res. 2016, 76, 7151–7159, doi:10.1158/0008-5472.CAN-16-1624.
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 isoform Δ133p53β in glioblastomas: An alternative to mutant p53 in promoting tumor development. J. Pathol. 2018, 246, 77–88, doi:10.1002/path.5111.
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 631, doi:10.1038/s41419-019-1861-1.
- Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C.G. Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts. Cancers 2020, 12, 769, doi:10.3390/cancers12030769.
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142, doi:10.1038/ncb1928.
- Arsic, N.; Ho-Pun-Cheung, A.; Evelyne, C.; Assenat, E.; Jarlier, M.; Anguille, C.; Colard, M.; Pezet, M.; Roux, P.; Gadea, G. The p53 isoform delta133p53ß regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS ONE 2017, 12, e0172125, doi:10.1371/journal.pone.0172125.
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254, doi:10.1038/s41467-017-02408-0.
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the ∆133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188, doi:10.3892/ijo.2013.1818.
- Nutthasirikul, N.; Hahnvajanawong, C.; Techasen, A.; Limpaiboon, T.; Leelayuwat, C.; Chau-In, S.; Jearanaikoon, P. Targeting the ∆133p53 isoform can restore chemosensitivity in 5-fluorouracil-resistant cholangiocarcinoma cells. Int. J. Oncol. 2015, 47, 2153–2164, doi:10.3892/ijo.2015.3188.
- Fragou, A.; Tzimagiorgis, G.; Karageorgopoulos, C.; Barbetakis, N.; Lazopoulos, A.; Papaioannou, M.; Haitoglou, C.; Kouidou, S. Increased Δ133p53 mRNA in lung carcinoma corresponds with reduction of p21 expression. Mol. Med. Rep. 2017, 15, 1455–1460, doi:10.3892/mmr.2017.6162.
- Tu, Q.; Gong, H.; Yuan, C.; Liu, G.; Huang, J.; Li, Z.; Luo, J. Δ133p53/FLp53 Predicts Poor Clinical Outcome in Esophageal Squamous Cell Carcinoma. Cancer Manag Res. 2020, 12, 7405–7417, doi:10.2147/CMAR.S263559.
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599, doi:10.1038/bjc.2011.433.
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244, doi:10.1038/s41598-019-41706-z.
- Knezović Florijan, M.; Ozretić, P.; Bujak, M.; Pezzè, L.; Ciribilli, Y.; Kaštelan, Ž.; Slade, N.; Hudolin, T. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. Urol. Oncol. 2019, 37, 578.e1–578.e10, doi:10.1016/j.urolonc.2019.03.007.
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. ELife 2016, 5, e14734, doi:10.7554/eLife.14734.
- Milićević, Z.; Bajić, V.; Živković, L.; Kasapović, J.; Andjelković, U.; Spremo-Potparević, B. Identification of p53 and its isoforms in human breast carcinoma cells. Sci. World J. 2014, 2014, 618698, doi:10.1155/2014/618698.
- Ozretić, P.; Hanžić, N.; Proust, B.; Sabol, M.; Trnski, D.; Radić, M.; Musani, V.; Ciribilli, Y.; Milas, I.; Puljiz, Z.; et al. Expression profiles of p53/p73, NME and GLI families in metastatic melanoma tissue and cell lines. Sci. Rep. 2019, 9, 12470, doi:10.1038/s41598-019-48882-y.
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369, doi:10.1038/cr.2015.22.
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. p53 isoform Δ133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 2016, 6, 37281, doi:10.1038/srep37281.
- Horikawa, I.; Park, K.-Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028, doi:10.1038/cdd.2017.48.
- Oh, L.; Hainaut, P.; Blanchet, S.; Ariffin, H. Expression of p53 N-terminal isoforms in B-cell precursor acute lymphoblastic leukemia and its correlation with clinicopathological profiles. BMC Cancer 2020, 20, 110, doi:10.1186/s12885-020-6599-8.
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Natl. Acad. Sci. USA 2012, 109, E2543–E2550, doi:10.1073/pnas.1205664109.
- Zhang, H.-M.; Sang, X.-G.; Wang, Y.-Z.; Cui, C.; Zhang, L.; Ji, W.-S. Role of Δ133p53 isoform in NF-κB inhibitor PDTC-mediated growth inhibition of MKN45 gastric cancer cells. World J. Gastroenterol. 2017, 23, 2716–2722, doi:10.3748/wjg.v23.i15.2716.
- Chambers, S.K.; Martinez, J.D. The significance of p53 isoform expression in serous ovarian cancer. Future Oncol. Lond. Engl. 2012, 8, 683–686, doi:10.2217/fon.12.60.

