 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Farzad Pakdel | + 1993 word(s) | 1993 | 2021-01-20 03:35:01 | | | |
| 2 | Dean Liu | -639 word(s) | 1354 | 2021-01-25 06:50:57 | | |
Video Upload Options
Breast cancer (BC) is the most common cancer among women worldwide. More than 70% of BC cases express estrogen receptor alpha (ERα), a central transcription factor that stimulates the proliferation of breast cancer cells, usually in the presence of estrogen.
1. Introduction
Millions of women develop breast cancer worldwide, representing a major health issue. Notably, breast cancer also exists in men but is very rare, accounting for fewer than one percent of cases. This disease generally arises from the proliferation of epithelial cells in the lobules or lactiferous ducts of the mammary gland and is a very heterogeneous malignancy. According to histopathological data, breast cancers are classified as lobular or ductal, in situ or invasive carcinomas preferentially colonizing bone, liver, lung or brain. In addition to histological grade and cancer stage determination, the development of molecular techniques has shed light on the heterogeneity of molecular profiles and gene expression across the types of breast cancer, resulting in more than 20 subtypes of breast carcinoma being characterized[1][2]. Molecular markers rely primarily on the expression of relevant receptors, including estrogen receptor alpha (ERα), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2). Various expressions of these receptors by breast cancer cells are correlated with different degrees of differentiation and aggressiveness of the tumor. Knowing these characteristics enables improved prognosis and selection of the most relevant therapy[3][4]. The simplest classification relies on three major subtypes: luminal, HER2-enriched, and triple-negative, as shown in Table 1[5].
Table 1. Breast cancer classification based on receptor expression (ER: estrogen receptor; PR: progesterone receptor; HER2: human epidermal growth factor receptor 2).
| Breast Cancer Type | Proportion | Biological Profile | Therapy of Choice |
|---|---|---|---|
| Luminal | Endocrine therapy | ||
| A | 60% | ERα+ PR+/− HER2− | |
| B | 10% | ERα+ PR+/− HER2+ | |
| HER2-enriched[6] | 20% | ERα+/− PR+/− HER2+ | Anti-HER2 therapy |
| Triple negative[7] | ERα− PR− HER2− | Chemotherapy | |
| Basal-like | 7% | + basal markers | |
| Non-basal-like | 3% | − basal markers |
The luminal type of breast cancer is divided into two subclasses: A and B. Both are characterized by the expression of ERα, but differ in terms of aggressiveness: luminal A breast cancers are usually low-grade, whereas luminal B cancers display overexpression of HER2, reduced ERα expression, and increased proliferation. Such breast cancers are predominant, especially luminal A, and convey a better prognosis because these tumors depend on estrogen for their growth, and specifically targeting estrogen through endocrine therapy to block its proliferative action, which is an effective strategy. As reviewed by Jensen and Jordan, the identification of the estrogen receptor and subsequent understanding of its implication in breast cancer paved the way for developing targeted therapy[8]. The selective estrogen receptor modulator (SERM) tamoxifen was first developed during the 1970s and became the standard of care for breast cancer, as it enabled the saving of many lives with fewer side effects than chemotherapy. Nevertheless, the use of tamoxifen presents some drawbacks, which led to the development of additional SERMs, as reviewed by Maximov et al.[9]. While SERMs are competitive inhibitors of ERα that prevent its activation by estrogens, another way to counteract ERα activity relies on using selective estrogen receptor downregulators (SERDs), whose binding to ERα results in the degradation of the receptor. Finally, another therapeutic approach aims to directly deprive the tumor of estrogen by ovariectomy or the use of aromatase inhibitors. The development of this class of agents began during the 1980s by Brodie and colleagues and had benefits for patients who do not respond to SERMs[10]. However, for 30–50% of ER-positive breast cancers, resistance to endocrine therapy occurs. For this large number of patients, the prognosis is worse, which raises real public health concerns[5].
2. ERα Activity
Estrogen receptors are nuclear receptors that mediate estrogen actions by regulating gene expression. Two highly homologous protein isoforms exist in vertebrates, ERα and ERβ, encoded by two independent genes. However, several primarily in vivo studies have shown that their activity differs in mammary gland development and nonreproductive tissue functioning, as well as in breast cancer pathogenesis [11]. This difference is in line with their differential tissue expression. Concerning breast cancer, clinicopathological data notably reveal that ERβ levels decrease during carcinogenesis[12][13]. The precise role of ERβ remains elusive; several lines of evidence confer it tumor suppressive activity, but further studies are needed to gain insights into the mechanisms involved[14].
Much more is known about ERα. Its activity is essential for the reproductive system, and its expression is increased in most breast cancers, which has led to extensive research to understand its regulatory role. ERα is a ligand-inducible transcriptional factor. After ligand binding and dimerization, ERα is recruited to the promoter region of the target genes, either by binding, directly targeting DNA sequences called EREs (estrogen responsive elements), or by protein/protein interactions with other transcriptional factors, such as AP1 or SP1. Recent development of chromatin immunoprecipitation of DNA coupled to high-throughput sequencing (ChIP-Seq) techniques has led to the identification of the ERα cistrome in mammary adenoma carcinoma cell lines, such as MCF-7, with 5000 to 10,000 estrogen receptor binding sites (ERBS), three quarters of which are EREs[15]. This results in the recruitment of numerous coactivators (members of the p160 family, CBP/P300, members of the SWI/SNF family, members of the mediator complex, etc.) via the AF1 and AF2 transactivation domains of ERα in an ordered, cyclic and combinatorial process, leading to transcriptional activation of target genes[16]. In addition to this activity at the genomic level, ERα also has nongenomic activity by interacting in the cytoplasm with cellular kinases that activate various signaling pathways, such as PI3K-AKT and Src-MAPK (Figure 1). These rapid actions of the receptor may ultimately result in the regulation of gene expression, highlighting the complex interrelationships between membrane and nuclear-induced events. All these modes of action ultimately lead to the regulation of cell fate, resulting in a balance between proliferation, differentiation and cell survival[11].
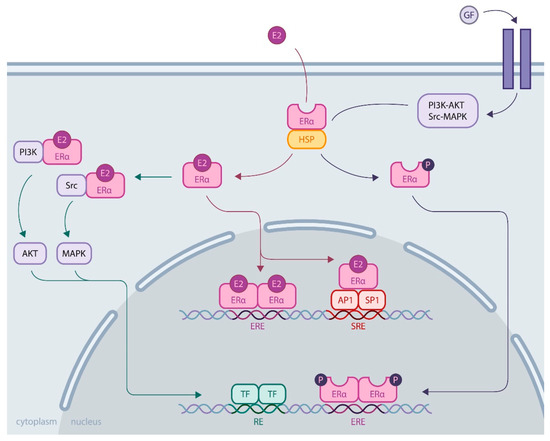
Figure 1. ERα activity through genomic and nongenomic actions. Genomic activity: ERα activation upon estrogen binding (E2) or after its phosphorylation by cellular kinases following growth factor (GF) receptor stimulation allows its release from heat shock proteins (HSPs). Then, ERα translocates into the nucleus, where it binds DNA by direct (through estrogen responsive elements, EREs) or indirect mechanisms (upon binding to other transcription factors, such as AP1 or SP1, that bind DNA through serum responsive elements, SREs). Nongenomic activity: Activated ERα interacts with cellular kinases (e.g., PI3K and Src), leading to signaling pathway stimulation involving AKT or MAPK, for example, eventually resulting in transcription factor (TF) activation. All these mechanisms induce transcriptional activation or repression of the regulation of cell fate.
3. Role of ERα in Breast Cancer
Many players are involved in breast carcinogenesis at the cellular level. Notable examples include tumor suppressor genes BRCA1/2, TP53 or PTEN undergoing loss-of-function mutations or decreased expression, receptor tyrosine kinases overexpression, such as EGFR, IGF1R or HER2, whose downstream signaling pathways promote cell proliferation and invasion (PI3K-AKT, RAS-MAPK, JNK) or overexpression of the oncogene c-Myc[17][18].
However, for most breast cancers, ER deregulation plays a major role, participating in aberrant cell proliferation and leading to tumor development. This deregulation of ERα function is due to multiple phenomena, implicating a shift in the balance of cofactors in favor of coactivators, the overexpression and function of growth factor receptors whose signaling pathways (MAPK, PI3K, etc.) result in ERα activation or alteration of ERα expression at the genetic level through epigenetic regulation[19][20][21]. ERα can also be mutated and become constitutively active, but it is not the primary source of breast cancer development. Instead, activating mutations are acquired following estrogen deprivation therapies as a resistance mechanism of tumor cells to escape hormonal control and promote cell proliferation through ligand-independent activation of ERα [22]. Breast cancer cells have evolved into many other strategies against endocrine therapies, as reviewed by Musgrove and Sutherland[23].
Recent developments in biophysical techniques have enabled us to gain insights into ERα conformational changes related to mutations, causing increasing interest for understanding of the ERα response to estrogen and antiestrogens. Whereas several ERα alterations have been reviewed elsewhere, such as gene amplification or translocation[24], as well as splice variants[22], this review aims to summarize the latest advances relative to the particular implication of ERα point mutations in endocrine resistance.
References
- Lakhani, S.; Ellis, I.; Schnitt, S.; Tan, P.; van de Vijver, M. WHO Classification of Tumours of the Breast, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2012; ISBN 978-92-832-2433-4.
- Vuong, D.; Simpson, P.T.; Green, B.; Cummings, M.C.; Lakhani, S.R. Molecular Classification of Breast Cancer. Virchows Arch. 2014, 465, 1–14.
- Bertucci, F.; Finetti, P.; Rougemont, J.; Charafe-Jauffret, E.; Cervera, N.; Tarpin, C.; Nguyen, C.; Xerri, L.; Houlgatte, R.; Jacquemier, J.; et al. Gene Expression Profiling Identifies Molecular Subtypes of Inflammatory Breast Cancer. Cancer Res. 2005, 65, 2170–2178.
- Gajulapalli, V.N.R.; Malisetty, V.L.; Chitta, S.K.; Manavathi, B. Oestrogen Receptor Negativity in Breast Cancer: A Cause or Consequence? Biosci. Rep. 2016, 36, e00432.
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine Resistance in Breast Cancer—An Overview and Update. Mol. Cell. Endocrinol. 2015, 418, 220–234.
- Costa, R.L.B.; Czerniecki, B.J. Clinical Development of Immunotherapies for HER2 + Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 10.
- Diana, A.; Carlino, F.; Franzese, E.; Oikonomidou, O.; Criscitiello, C.; De Vita, F.; Ciardiello, F.; Orditura, M. Early Triple Negative Breast Cancer: Conventional Treatment and Emerging Therapeutic Landscapes. Cancers 2020, 12, 819.
- Jensen, E.V.; Jordan, V.C. The Estrogen Receptor: A Model for Molecular Medicine. Clin. Cancer Res. 2003, 9, 1980–1989.
- Maximov, P.; Lee, T.; Jordan, V. The Discovery and Development of Selective Estrogen Receptor Modulators (SERMs) for Clinical Practice. CCP 2013, 8, 135–155.
- Jordan, V.C.; Brodie, A.M.H. Development and Evolution of Therapies Targeted to the Estrogen Receptor for the Treatment and Prevention of Breast Cancer. Steroids 2007, 72, 7–25.
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087.
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor Beta in Breast Cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672.
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.-Å. Differential Expression of Estrogen Receptor α, Β1, and Β2 in Lobular and Ductal Breast Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938.
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-Canonical Estrogen Signaling in Endocrine Resistance. Front. Endocrinol. 2019, 10, 708.
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-Wide Analysis of Estrogen Receptor α DNA Binding and Tethering Mechanisms Identifies Runx1 as a Novel Tethering Factor in Receptor-Mediated Transcriptional Activation. MCB 2010, 30, 3943–3955.
- Métivier, R.; Penot, G.; Hübner, M.R.; Reid, G.; Brand, H.; Koš, M.; Gannon, F. Estrogen Receptor-α Directs Ordered, Cyclical, and Combinatorial Recruitment of Cofactors on a Natural Target Promoter. Cell 2003, 115, 751–763.
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397.
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53.
- Murphy, L.; Cherlet, T.; Lewis, A.; Banu, Y.; Watson, P. New Insights into Estrogen Receptor Function in Human Breast Cancer. Ann. Med. 2003, 35, 614–631.
- Manavathi, B.; Dey, O.; Gajulapalli, V.N.R.; Bhatia, R.S.; Bugide, S.; Kumar, R. Derailed Estrogen Signaling and Breast Cancer: An Authentic Couple. Endocr. Rev. 2013, 34, 1–32.
- Kerdivel, G.; Flouriot, G.; Pakdel, F. Modulation of Estrogen Receptor Alpha Activity and Expression During Breast Cancer Progression. In Vitamins & Hormones; Elsevier: Cambridge, MA, USA, 2013; Volume 93, pp. 135–160. ISBN 978-0-12-416673-8.
- Barone, I.; Brusco, L.; Fuqua, S.A.W. Estrogen Receptor Mutations and Changes in Downstream Gene Expression and Signaling. Clin. Cancer Res. 2010, 16, 2702–2708.
- Musgrove, E.A.; Sutherland, R.L. Biological Determinants of Endocrine Resistance in Breast Cancer. Nat. Rev. Cancer 2009, 9, 631–643.
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of Aromatase Inhibitor Resistance. Nat. Rev. Cancer 2015, 15, 261–275.



