 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Haitang Yang | + 2654 word(s) | 2654 | 2021-01-11 07:36:38 | | | |
| 2 | Vivi Li | Meta information modification | 2654 | 2021-01-21 07:26:18 | | |
Video Upload Options
Regulatory networks controlling cellular plasticity, important during early development, can re-emerge after tissue injury and premalignant transformation. One such regulatory molecule is the cell surface ectoenzyme ecto-5′-nucleotidase that hydrolyzes the conversion of extracellular adenosine monophosphate to adenosine (eADO). Ecto-5′-nucleotidase (NT5E) or cluster of differentiation 73 (CD73), is an enzyme that is encoded by NT5E in humans. In normal tissue, CD73-mediated generation of eADO has important pleiotropic functions ranging from the promotion of cell growth and survival, to potent immunosuppression mediated through purinergic G protein-coupled adenosine receptors. Importantly, tumors also utilize several mechanisms mediated by CD73 to resist therapeutics and in particular, evade the host immune system, leading to undesired resistance to targeted therapy and immunotherapy. Tumor cell CD73 upregulation is associated with worse clinical outcomes in a variety of cancers. Emerging evidence indicates a link between tumor cell stemness with a limited host anti-tumor immune response.
1. Introduction
Cellular plasticity represents a broad phenomenon whereby cells change their identity or state representing an important event in early development enabling proper tissue morphogenesis [1]. One of the best-studied examples of cellular plasticity is the epithelial-to-mesenchymal transition (EMT) whereby epithelial cells take on characteristics of mesenchymal cells while becoming more motile [2], which is essential during gastrulation and neural crest formation [3]. Under homeostatic conditions in adult tissues, cellular identity was originally believed to be hard-wired and non-malleable. However, it is now apparent that during periods of chronic perturbation, cellular identity is more pliant whereby cells within stressed tissues transition between cellular states, which is part of a natural adaptive process during wound repair and tissue regeneration [4].
Cellular plasticity is also recognized as a hallmark of cancer [5]. Importantly, poorly differentiated tumors reflected in the histological grade and a product of an EMT program are more prone to metastatic spread and poor prognosis, also are enriched in gene signatures associated with dedifferentiation and stemness [6][7]. Our understanding of the drivers of cellular plasticity in cancer is not well understood. While wide-scale cell-autonomous genomic alterations (e.g., coding mutations, chromosomal alterations) have long been recognized as a basis for tumor initiation [8], there is emerging evidence suggesting that non-cell-autonomous processes involving adaptive properties of the microenvironment also are critical features that contribute to tumor plasticity [9].
Cluster of differentiation 73 (CD73; encoded by NT5E [ecto-5′-nucleotidase]) molecule is a membrane-bound ecto-5′-nucleotidase ubiquitously expressed throughout the body [10] and is the major enzyme responsible for the generation of extracellular adenosine (eADO) via enzymatic dephosphorylation of nucleotide adenosine 5′-monophosphate (AMP) [11]. Other AMP-hydrolyzing enzymes can also be involved in the generation of eADO such as CD39 and ectonucleotide phosphodiesterase 1 (ENPP1), which have been covered by other excellent reviews [12][13]. CD73 has been shown to impart particularly important cell-type-specific functions involved in regulating tissue homeostasis. The physiological functions regulated by CD73-mediated generation of eADO are coupled to the interaction with seven-transmembrane domain, G-protein-coupled receptors adenosine receptor A1 (ADORA1), ADORA2A, ADORA2B, and ADORA3, each possessing a unique binding affinity and signal transduction mechanism (for a review see Olah and Stiles [14]). Under normal physiological conditions, eADO is present at low concentrations in tissues, including the brain. During periods of heightened metabolic stress such as that occurs during prolonged inflammation, eADO levels are greatly increased by enhanced 5′-nucleotidase CD73 to regulate immunity and inflammation, which is a necessary response to ensure tissue repair following injury [15].
There is emerging evidence suggesting that the CD73-adenosinergic signaling pathway is hijacked by tumors that arise because of prolonged periods of inflammation, resulting in suppression of immune-mediated disease control of tumors or limitations for resources and cell competitions due to a limited supply of oxygen [16]. Hypoxia and transforming growth factor-beta 1 (TGF-β1) represent two of the most prominent extrinsic features of the microenvironment in solid tumors that are responsible for initiating a transcriptional program triggering cellular plasticity in carcinoma cells in the form of developmental differentiation programs EMT and stemness [17]. Importantly, these changes in the tumor microenvironment (TME) also serve as drivers of CD73 expression, so it is not surprising that there is a possible link between tumorigenesis and increased CD73 expression. In this review, we will focus on the functional role of CD73 in solid tumors and its correlation with signatures of cellular plasticity (mRNA and epigenetic signatures together with an EMT signature) and host immune response, with a particular focus on solid tumors. In addition, we will touch upon the association between CD73 and metastasis, which is responsible for over 90% of cancer deaths. Finally, we will discuss the current state of drug discovery and drug development efforts focusing on CD73 and the potential of targeting the CD73-adenosinergic signaling pathway by drug repurposing.
2. CD73 and Human Cancer
Several recent studies have provided evidence that overexpression of CD73 is associated with poor patient outcomes across diverse cancer cohorts [18][19][20][21]. Gene expression analysis of the pan-cancer cohort from the public The Cancer Genome Atlas (TCGA) reveals that mRNA expression of NT5E is highly heterogeneous across different cancer types. For example, the most abundant expression of NT5E was observed in thyroid carcinoma (THCA), glioblastoma (GBM), sarcoma (SARC), and minimal in blood cancer (LAML, acute myeloid leukemia) (Figure 1A). Compared to matched normal tissue, NT5E is highly upregulated in the majority of solid cancer types (Figure 1B). Interestingly, this is not the case for tumors of the genitourinary system, e.g., cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), ovarian serous cystadenocarcinoma (OV), testicular germ cell tumors (TGCT), uterine corpus endometrial carcinoma (UCEC), uterine carcinosarcoma (UCS), bladder urothelial carcinoma (BLCA), kidney chromophobe (KICH) and prostate adenocarcinoma (PRAD). These observations may be due to the common embryological origin (the intermediate mesoderm) of the reproductive and urinary systems. Consistent with previous reports demonstrating the relationship between CD73 and patient survival, we demonstrate that high expression of NT5E predicts poor patient survival in the majority of TCGA solid tumors that have overexpressed NT5E/CD73 relative to paired normal tissue (Figure 1C). Collectively, the evidence provided here suggests the critical role of CD73 in a subset of non-genitourinary system-derived solid cancers and prioritizes CD73 as a potential therapeutic target in cancer.
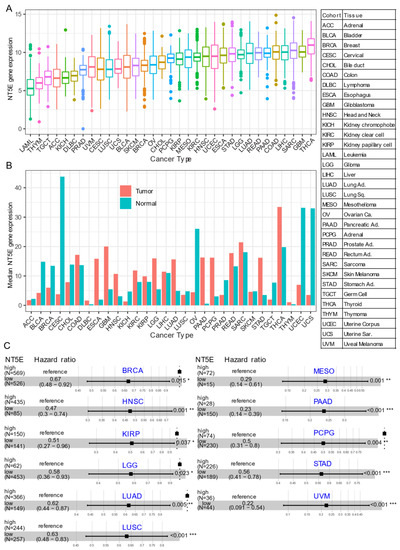
Figure 1. CD73 in human cancers. (A) NT5E (encoding CD73) gene expression across human solid tumors. Data were downloaded and reanalyzed from TCGA (The Cancer Genome Atlas Program) pan-cancer cohort. (B) Bar plot showing the NT5E gene expression in tumor compared with the matched normal tissue. The height of the bar represents the median expression of the indicated tumor type (red) or normal tissue (blue). (C) Forest blots showing the Cox proportional-hazards model-based survival analysis of cancer patients stratified by the gene expression of NT5E across the TCGA pan-solid cancer cohort. Only significant (p < 0.05) results were presented. The “high” and “low” expression groups were stratified by the optimal cutoff value using “survminer” and “survival” packages in R software. N, the total number in each group. Scale line indicates the 95% confidence interval for effect estimate for each survival-influencing factor with the hazard ratio showing to the right. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; UCS, uterine carcinosarcoma; UCEC, uterine corpus endometrial carcinoma; UVM, uveal melanoma. Ca., carcinoma; Ad., adenocarcinoma; Sq., squamous; Sa., sarcoma. * p < 0.05, ** p < 0.01, *** p < 0.001. The detailed information about the bioinformatic analysis can be found in Appendix A.
3. CD73 and Tumor Immune Microenvironment
Although cancer has primarily been studied as a cell-intrinsic disease, it is widely accepted that the tumor microenvironment (TME) plays an essential role in regulating plasticity. Along these lines, Malta et al. demonstrated that tumor types with higher stemness indices are correlated with reduced immune infiltration and PD-L1 expression at the protein level [22]. CD73 is an emerging immune checkpoint in modulating cancer progression via conversion of immunostimulatory eATP into immunosuppressive eADO [23][24]. As described above, CD73 nucleotidase activity promoting an immunosuppressive TME represents an ideal target to enhance immunotherapies in cancer, which to date are underwhelming in the majority of patients [25][26][27].
Comprehensive profiling of over 10,000 tumors across 33 diverse cancer types from TCGA pan-solid cancer cohort using a multi-omics approach uncovered six (C1–C6) immune subtypes [28]. This work provides a key resource for understanding tumor-immune interactions. The lymphocyte-depleted (immune C4) and TGF-beta dominant (immune C6) subtypes were marked by the worst prognosis and their immune makeup was consistent with an immunosuppressed TME. Accumulating evidence points towards CD73 shaping the TME [15][16]. Further exploration revealed that the Immunologically Quiet (Immune C5) and TGF−beta dominant (immune C6) immune subtypes are highly enriched in tumors with high NT5E expression compared with low NT5E expression (Figure 4A). To gain a clearer picture of the immune infiltrates across the tumor subtypes which may be of potential relevance to cancer immunology, we utilized TIMER, an algorithm that provides information regarding proportions of immune cell types by multiple immune deconvolution methods [29]. Focusing primarily on T cells, we demonstrate that T cell regulatory (Tregs)_QUANTISEQ fraction is positively correlated with CD73 gene expression across a wide number of tumor types including BRCA and THCA. In addition, CD73 gene expression displays a broadly positive correlation with the T cell CD8+_TIMER and multiple CD4+ fractions (Figure 4B). This pan-cancer dataset suggests a mixture of suppressive and activated immune infiltrates together with high CD73 gene expression. For example, in HNSC a negative correlation between CD73 and CD8+ T cell infiltration, including HPV-positive patients was observed (Figure 4B). This observation supports a recent study from Watermann et al. [30] demonstrating that recurrent HNSC has an immunosuppressive tumor microenvironment with significant depletion of CD8 tumor-infiltrating lymphocytes (TILs), which was more pronounced following adjuvant chemo-radiotherapy. Therefore, these tumors might be less susceptible to respond to ICIs due to the lack of infiltrating CD8+ CTLs. Whether anti-CD73 treatment can turn an immunosuppressive cold tumor-like HNSC into a hot tumor awaits further investigation. It is important to note that CD73-mediated production of immunosuppressive adenosine has also been described for tumor-infiltrating B cells [31] and NK cells [32] whereby tumors can hijack both B cell- and NK cell-mediated suppression of activated T cells to escape immunity.
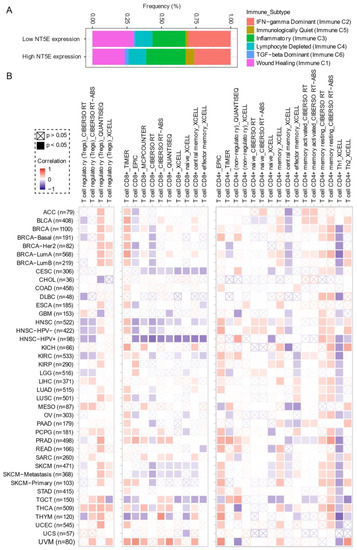
Figure 4. CD73 and tumor immune microenvironment. (A) Percentage (NT5E expression low vs. high) of immune subtype models (C1–C6) across TCGA (The Cancer Genome Atlas) pan-cancer cohort. The genes contained in each signature were evaluated using model-based clustering by p the “mclust” R package. Each sample was finally to be grouped based on its predominance with the C1–C6 signature. The immune subtype models were based on Thorsson V et al. Immunity. 2018 (see the methods in Appendix A). (B) Systematic correlation analysis of immune infiltrates (Tregs [left], CD8+ [middle], CD4+ [right]) with gene expression of NT5E across TCGA pan-cancer cohort. The number of patients was shown in parenthesis. Data were downloaded from TIMER (version 2.0), a comprehensive resource for systematic analysis of immune infiltrates across diverse cancer types (http://timer.comp-genomics.org/) (Ref. [29]). The red color indicates a positive correlation, while the blue color represents a negative correlation. The detailed information about the bioinformatic analysis can be found in Appendix A.
4. CD73 and Drug Repurposing
The above evidence highlights the importance of blocking the CD73-adenosinergic signaling pathway in the treatment of cancer. Currently, there is an intense effort underway developing novel monoclonal antibodies or selective small molecules targeting both CD73 or P1 adenosinergic receptors (A2aRA, A2aRB) for cancer and we refer the reader to recent in-depth reviews covering this [16][33]. Despite the promise of CD73 as a therapeutic target for cancer treatment, there are currently no clinically approved CD73 inhibitors for the treatment of cancer. One of the main reasons behind this is the lack of biomarkers of CD73-adenosine rich tumors to guide precise clinical management, which may lead to heterogeneous treatment responses and rapid development of resistance to CD73 targeted therapy. Preclinical mouse models and human studies confirm the link between hypoxia and CD73-adenosinergic rich tumors and chemoresistance [34]. Therefore, a bioinformatics biomarker-guided stratification that will identify patients most likely to benefit from CD73 targeted therapy alone or in combination with chemotherapy, targeted, or immunotherapy is needed.
Given the unavailability of clinically approved CD73 inhibitors for cancer, drug repurposing, which is intended to find new uses for clinically approved drugs [35], may hold promise as an alternative and cost-effective way of targeting the CD73-adenosinergic signaling pathway. The advantage of this approach is that the existing pharmacokinetic/pharmacodynamic, as well as safety profiles have already been established [35]. To systematically investigate potential inhibitor candidates that can modulate the efficacy of CD73 inhibitors, drug sensitivity profiles of hundreds of compounds (n = 481) were correlated with CD73 mRNA level across a panel of solid cancer cell lines (n = 659) [36]. Of note, a negative correlation means that cancer cells with a high expression of CD73 have a lower AUC (area under the curve) value in response to the indicated inhibitors representing a sensitive drug compound (Figure 5A). In contrast, a positive correlation indicates that solid cancer cells with a high expression of CD73 are more resistant to the corresponding drug compounds. Intriguingly, we observed that dasatinib (brand name Sprycel), a clinically approved inhibitor selectively targeting BCR-ABL/SRC used in the treatment of chronic myeloid leukemia and acute lymphoblastic leukemia, is the only compound whose AUC value significantly negatively correlates with the expression of CD73. Conversely, several pan-HDAC inhibitors appear as the top whose AUC significantly positively correlated drug compounds. Therefore, cancer cells with enhanced CD73 activity might be sensitive to dasatinib treatment, but resistant to pan-HDAC inhibitors. Regarding this, we and others have previously shown that dasatinib not only inhibits cancer cells but also modulates the tumor immune microenvironment and enhances the efficacy of ICIs [37][38][39]. Others have shown that dasatinib effectively blocks TGFβ-induced expression of transcription factors promoting EMT and may be a novel therapeutic option in pancreatic and prostate cancer [40] and pulmonary sarcomatoid carcinoma, a rare and deadly form of NSCLC [41].
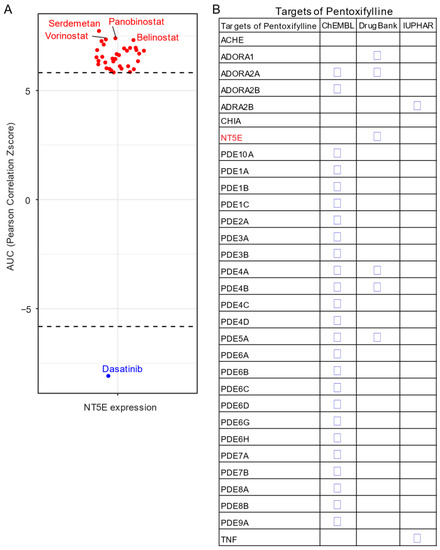
Figure 5. Drug repurposing for CD73 targeted therapy. (A) Integrated correlation analysis of NT5E gene expression with drug (n = 481) response profiles (reflected by Z-score normalized area under the curve [AUC] value) across solid cancer cell lines (n = 659). Red dots indicate drugs whose AUC value significantly (adj. p < 0.05) positively correlates with NT5E gene expression, while the blue represent the significantly negatively correlated ones. The drug response data were downloaded from a previously published study (see the methods in Appendix A). (B) Drug repurposing identifying NT5E as one target of Pentoxifylline. Data were downloaded from ReframeDB database (https://reframedb.org/). The detailed information about the bioinformatic analysis can be found in Appendix A.
Based on a public drug repurposing database ReframeDB (https://reframedb.org/), we have found that the synthetic dimethylxanthine derivative pentoxifylline (PTX), clinically approved for the treatment of peripheral vascular diseases and osteoradionecrosis, as well as the management of cerebrovascular insufficiency [42][43] also targets the CD73-adenosinergic signaling pathway (Figure 5B). Originally identified as a nonselective phosphodiesterase inhibitor (PDE4B, PDE4A, PDE5A), PTX also has additional anti-inflammatory, immunomodulatory and bronchodilatory effects due to its ability to act as a non-selective adenosine receptor antagonist (A1, ADORA1 and A2a, ADORA2A) and NT5E/CD73 inhibitor [44]. CD73 is upregulated following radiotherapy and is linked to radiation-induced tissue injury [45] and PTX shows protective effects against radiotherapy-induced lung toxicity in both breast and lung cancer [46]. Blocking radiotherapy-induced CD73 upregulation promotes host-mediated immune rejection of tumors [47]. Collectively, repurposing of well-known clinically approved drugs targeting CD73-adenosine together in combination with dasatinib and/or PTX with ICIs may represent an ideal combination therapy in solid tumors with an extensive desmoplastic stroma, which warrants further investigation.
References
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851.
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45.
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486.
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27.
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56.
- Zheng, H.; Song, K.; Fu, Y.; You, T.; Yang, J.; Guo, W.; Wang, K.; Jin, L.; Gu, Y.; Qi, L.; et al. An absolute human stemness index associated with oncogenic dedifferentiation. Brief. Bioinform. 2020.
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507.
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628.
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376.
- Minor, M.; Alcedo, K.P.; Battaglia, R.A.; Snider, N.T. Cell type-and tissue-specific functions of ecto-5’-nucleotidase (CD73). Am. J. Physiol. Cell Physiol. 2019, 317, C1079–C1092.
- Robson, S.C.; Sevigny, J.; Zimmermann, H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. 2006, 2, 409–430.
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347.
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020.
- Olah, M.E.; Stiles, G.L. Adenosine receptor subtypes: Characterization and therapeutic regulation. Annu. Rev. Pharm. Toxicol. 1995, 35, 581–606.
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367.
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629.
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629.
- Jeong, Y.M.; Cho, H.; Kim, T.M.; Kim, Y.; Jeon, S.; Bychkov, A.; Jung, C.K. CD73 Overexpression Promotes Progression and Recurrence of Papillary Thyroid Carcinoma. Cancers 2020, 12, 3042.
- Park, L.C.; Rhee, K.; Kim, W.B.; Cho, A.; Song, J.; Anker, J.F.; Oh, M.; Bais, P.; Namburi, S.; Chuang, J.; et al. Immunologic and clinical implications of CD73 expression in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 12050.
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.; Haibe-Kains, B.; Karn, T.; et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015, 75, 4494–4503.
- Leclerc, B.G.; Charlebois, R.; Chouinard, G.; Allard, B.; Pommey, S.; Saad, F.; Stagg, J. CD73 Expression is an Independent Prognostic Factor in Prostate Cancer. Clin. Cancer Res. 2016, 22, 158–166.
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.
- Massaia, M.; Pileri, A.; Boccadoro, M.; Bianchi, A.; Palumbo, A.; Dianzani, U. The generation of alloreactive cytotoxic T lymphocytes requires the expression of ecto-5’nucleotidase activity. J. Immunol. 1988, 141, 3768–3775.
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237.
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900.
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382.
- Yegutkin, G.G.; Marttila-Ichihara, F.; Karikoski, M.; Niemela, J.; Laurila, J.P.; Elima, K.; Jalkanen, S.; Salmi, M. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur. J. Immunol. 2011, 41, 1231–1241.
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514.
- Watermann, C.; Pasternack, H.; Idel, C.; Ribbat-Idel, J.; Bragelmann, J.; Kuppler, P.; Offermann, A.; Jonigk, D.; Kuhnel, M.; Schrock, A.; et al. Recurrent HNSCC harbor an immunosuppressive tumor immune microenvironment suggesting successful tumor immune evasion. Clin. Cancer Res. 2020.
- Saze, Z.; Schuler, P.J.; Hong, C.S.; Cheng, D.; Jackson, E.K.; Whiteside, T.L. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood 2013, 122, 9–18.
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.P.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J. Clin. Investig. 2020, 130, 1185–1198.
- Jeffrey, J.L.; Lawson, K.V.; Powers, J.P. Targeting Metabolism of Extracellular Nucleotides via Inhibition of Ectonucleotidases CD73 and CD39. J. Med. Chem. 2020.
- Sitkovsky, M.V. Lessons from the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 16–19.
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58.
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116.
- Yang, H.; Xu, D.; Yang, Z.; Yao, F.; Zhao, H.; Schmid, R.A.; Peng, R.W. Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma. Cancers 2020, 12, 2310.
- Chen, R.; Lee, W.C.; Fujimoto, J.; Li, J.; Hu, X.; Mehran, R.; Rice, D.; Swisher, S.G.; Sepesi, B.; Tran, H.T.; et al. Evolution of genomic and T cell repertoire heterogeneity of malignant pleural mesothelioma under dasatinib treatment. Clin. Cancer Res. 2020.
- Tu, M.M.; Lee, F.Y.F.; Jones, R.T.; Kimball, A.K.; Saravia, E.; Graziano, R.F.; Coleman, B.; Menard, K.; Yan, J.; Michaud, E.; et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci. Adv. 2019, 5, eaav2437.
- Moro, L.; Simoneschi, D.; Kurz, E.; Arbini, A.A.; Jang, S.; Guaragnella, N.; Giannattasio, S.; Wang, W.; Chen, Y.A.; Pires, G.; et al. Epigenetic silencing of the ubiquitin ligase subunit FBXL7 impairs c-SRC degradation and promotes epithelial-to-mesenchymal transition and metastasis. Nat. Cell. Biol. 2020, 22, 1130–1142.
- Manzotti, G.; Torricelli, F.; Benedetta, D.; Lococo, F.; Sancisi, V.; Rossi, G.; Piana, S.; Ciarrocchi, A. An Epithelial-to-Mesenchymal Transcriptional Switch Triggers Evolution of Pulmonary Sarcomatoid Carcinoma (PSC) and Identifies Dasatinib as New Therapeutic Option. Clin. Cancer Res. 2019, 25, 2348–2360.
- Ward, A.; Clissold, S.P. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 1987, 34, 50–97.
- Kolokythas, A.; Rasmussen, J.T.; Reardon, J.; Feng, C. Management of osteoradionecrosis of the jaws with pentoxifylline-tocopherol: A systematic review of the literature and meta-analysis. Int. J. Oral Maxillofac. Surg. 2019, 48, 173–180.
- Marques, L.J.; Zheng, L.; Poulakis, N.; Guzman, J.; Costabel, U. Pentoxifylline inhibits TNF-alpha production from human alveolar macrophages. Am. J. Respir. Crit. Care Med. 1999, 159, 508–511.
- De Leve, S.; Wirsdorfer, F.; Jendrossek, V. The CD73/Ado System-A New Player in RT Induced Adverse Late Effects. Cancers 2019, 11, 1578.
- Ozturk, B.; Egehan, I.; Atavci, S.; Kitapci, M. Pentoxifylline in prevention of radiation-induced lung toxicity in patients with breast and lung cancer: A double-blind randomized trial. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 213–219.
- Wennerberg, E.; Spada, S.; Rudqvist, N.P.; Lhuillier, C.; Gruber, S.; Chen, Q.; Zhang, F.; Zhou, X.K.; Gross, S.S.; Formenti, S.C.; et al. CD73 Blockade Promotes Dendritic Cell Infiltration of Irradiated Tumors and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 465–478.

