 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nuno Osório | + 4124 word(s) | 4124 | 2021-01-19 09:45:38 | | | |
| 2 | Rita Xu | -2163 word(s) | 1961 | 2021-01-20 09:08:52 | | |
Video Upload Options
This entry reviews the genetic evolution and adaptation to the host environment of M. tuberculosis and HIV-1. It provides an overview of the latest developments in the knowledge about the genetic diversity of these devastating intracellular human pathogens and its impact on the host immune system, virulence, drug resistance propensity and other relevant aspects. We also proposed a novel topic in the literature regarding the use of single cell "omics" to study the interacting evolutionary dynamics of M. tuberculosis and HIV-1 in co-infection, at the cellular niche of monocytes/macrophages.
1. Introduction
Tuberculosis (TB), a disease caused by Mycobacterium tuberculosis infection, is one of the top ten leading causes of death worldwide [1][2]. Among the estimated 1.4 million TB deaths in 2019, 208,000 individuals were co-infected with the Human Immunodeficiency Virus (HIV), representing approximately 30% of the Acquired Immunodeficiency Syndrome (AIDS) related deaths (from a total of 690,000 [3]). Indeed, TB represents the leading cause of morbidity and mortality among AIDS patients [1][3][4]. Several efforts have been made to prevent transmission and treat the viral infection, relying on antiretroviral therapy (ART), which contributed to the avoidance of 12.1 million deaths due to AIDS related illnesses between the years 2010 and 2019 [3]. Regarding M. tuberculosis/HIV co-infection, the combined effect of ART and TB treatment prevented 11 million deaths between 2000 and 2019 [1].
According to the World Health Organization (WHO) in 2019, the risk of developing TB was 15 to 21 times higher in people infected with HIV. Within the same host, HIV and M. tuberculosis present a synergetic relationship, leading to a faster deterioration of the immune system and, consequently, to reduced life expectancy [5]. In fact, studies have shown that low CD4+ T cell counts in HIV-infected patients relate with a higher probability of developing active TB [4][6][7][8], while individuals undergoing ART therapy are less prone to develop TB [4].
One of the biggest challenges concerning TB treatment is the existence of drug-resistant M. tuberculosis strains. In 2019, approximately 500,000 cases of resistance to rifampicin, the most effective first-line drug, were reported, from which 78% presented multidrug-resistance (MDR) [1]. Interestingly, transmission of drug-resistant strains is considered the main contributor to this phenomenon, rather than genetic alterations of the bacterial genome arising de novo within infected individuals [9].
Contrasting with several other human pathogens, M. tuberculosis generates smaller genetic diversity levels within the host and at the transmission chain levels [10][11][12]. Still, two M. tuberculosis strains can differ in thousands of single nucleotide polymorphisms (SPNs) and associate with different clinically relevant phenotypes [11][13][14][15][16][17]. In addition, analysis of M. tuberculosis genomes revealed a biased geographic distribution for the phylogenetic lineages of the pathogen, supporting a possible adaptation to specific host populations [13][18][19][20][21][22][23].
On the other hand, HIV-1 presents extremely high genetic variability at the intrahost and population levels [24][25]. HIV-1 subtypes and recombinant forms have a dissimilar global distribution [26][27], and present distinct cell tropism, viral fitness and plasma viral loads, which may play a role on the infection dynamics, disease progression and in the response to ART [28][29]. Moreover, HIV-1 genetic diversity is behind the emergence of drug-resistant viruses and underlies the biggest challenges to find an effective treatment or cure to HIV-1 infection [30].
2. Genetic Diversity of M. Tuberculosis
2.1. M. tuberculosis Lineages: Origin and Host-Pathogen Associations
The Mycobacterium tuberculosis complex (MTBC) is composed of obligate pathogens with no known environmental reservoir [20][21][31]. Mycobacterium spp. from the MTBC share high levels of DNA sequence identity, which leads to controversial taxonomy and nomenclature [32][33]. Despite this, members of the MTBC differ widely in host range and geographic distribution [14][18][19][20][31][32]. There is a clear interspecies difference in the ability to cause active disease, with animal-adapted bacteria such as M. bovis or M. orygis being isolated occasionally in humans and associated with extra-pulmonary forms of the disease [20][31][34]. On the other hand, although the human pathogen M. tuberculosis has been occasionally isolated in other animals [35], associated pathology in the lungs and lymph nodes of cattle was found to be very low comparatively to M. bovis [36].
Human-adapted MTBC bacteria are divided into at least nine distinct lineages, as determined by whole-genome phylogenetic analysis using clinical isolates from several world regions [13][37][38]. These lineages are thought to have originated several millennia ago in the Neolithic period [39], with their phylogeographic pattern subsequently defined by sociodemographic events [13][22][40][41] and biological factors [10][15][32][42][43][44]. The “ancient” lineages 1, 5, 6 and the newly proposed lineage 9 (based on five genomes isolated from East African countries [38]) were considered the first to split from the common ancestor, while the “modern” lineages (lineages 2, 3 and 4) diverged later. Lineage 1 strains are more abundant in East Africa, Southeast Asia, South Asia and Oceania; lineage 2 in Eastern Asia (e.g., China); lineage 3 in East Africa, Southwest Asia, Central and South Asia; lineage 4 in Europe, Africa and America; lineages 5 and 6 are typically restricted to West Africa and lineage 7 was only isolated in Northern Ethiopia (Figure 1 and [13][45][46]). Additionally, two M. tuberculosis genomes from Rwanda and Uganda highlighted the existence of a sister clade to the MTBC (i.e., with an immediate common ancestor), which was named Lineage 8 [37]. The geographic distribution of these phylogenetic lineages across the globe is not likely to be a mere consequence of founder effect. Instead, it seems to be partially driven by the interaction with specific human populations [13][20][21]. Strikingly, the phylogeographic profile of M. tuberculosis is maintained even in highly urbanized settings, as demonstrated for San Francisco (USA). In this metropolis, the active TB cases in immigrant communities were predominantly caused by strains belonging to lineages that were characteristically abundant in the patient’s country of origin, rather than the highly prevalent lineage 4 strains [18][47]. On a similar note, the predominant sublineages in bordering areas of Southeast Asia were found to be different, contrasting with the high level of interregional flows of people [48]. This fact suggests that some level of host–pathogen co-adaptation exists and is important to the development of active TB. M. tuberculosis lineages are also substantially different in terms of human host population range, which might reflect on the relative success to cause active disease. Indeed, lineages 2 and 4 cover considerably more territory than the remaining lineages (Figure 1). Moreover, lineage 4 was isolated in patients from all inhabited continents. Nevertheless, certain sublineages of lineage 4 are more geographically restricted than others [23]. In addition, robust phylogenetic dating revealed the existence of these “specialist” sublineages over many centuries [46], demonstrating that there was enough time for a more global spread in light of globalization and airborne transmission. Interestingly, “specialist” sublineages presented lower levels of sequence diversity in experimentally validated T cell epitopes [23], indicating a possible role for human adaptive immune system in the definition of host-pathogen combinations that can more likely result in active TB after infection [23].
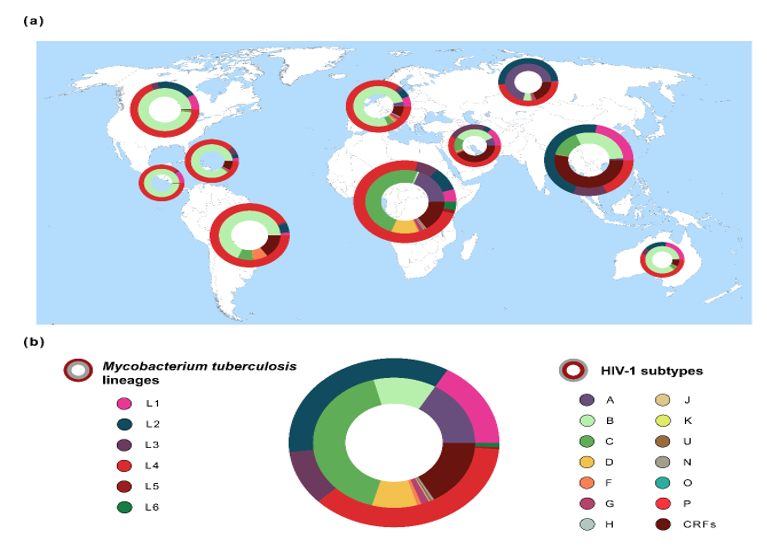
Figure 1. Global distribution of the genetic diversity of Mycobacterium tuberculosis and Human Immunodeficiency Virus typ1 1 (HIV-1). (a) Frequencies of M. tuberculosis lineages (outer ring/circle) and HIV-1 subtypes (inner ring/circle) across 10 geographic regions. Pie chart size is related with both diseases’ prevalence in each region [1][3]. Geographic regions are according to Los Alamos HIV database (http://www.hiv.lanl.gov/) [25]. M. tuberculosis spoligotype clades (n = 64,555 sequences) were extracted from the SITVIT2 database [49] and converted to Mycobacterium tuberculosis complex (MTBC) lineages 1–6 [45][50][51]. HIV-1 data (n = 815,431 sequences) were obtained from Los Alamos HIV database [25]. (b) Global frequency of M. tuberculosis lineages and HIV-1 subtypes. The estimated number of people living with TB or HIV-1 was obtained from the World Health Organization (WHO) [1] or from the Joint United Nations Programme on HIV/AIDS (UNAIDS) [3].
2.2. Biomedical Implication of M. tuberculosis Genetic Diversity
The population structure of M. tuberculosis was shown to influence several biomedically relevant aspects, such as transmissibility, disease severity, drug resistance and immune response (as extensively covered in previous reviews [10][15]). Here, we describe evidence of the interaction of M. tuberculosis with distinct human genetic backgrounds and HIV-1.
Transmissibility of a pathogen might ultimately dictate its prevalence among host populations. The transmission potential of “modern” MTBC lineage strains has a tendency to be higher than in their “ancient” partners [15][52]. Increases in relative prevalence have been consistently reported in several world regions for Lineage 2 [15][53][54][55]. In agreement, three quarters of the transmission clusters in Ho Chi Minh City (Vietnam) involved recently introduced lineage 2 strains [54]. Importantly, lineage 2 strains progressed to active disease in younger people and in shorter periods of time, while the endemic lineage 1 led to increased chance of reactivation from latent TB and in older people [54]. Another study demonstrated the ability of the Beijing sublineage of lineage 2 (L2-Beijing) to spread to Africa, following a series of multiple and timely independent events [55]. This suggests that the introduction of L2-Beijing was not solely due to its increased mutation rate, previously found to augment drug resistance acquisition in vitro [56]. Overall, both host and bacterial factors might be involved in the colonization of different ecological niches by M. tuberculosis lineages. One interesting approach is to dissect this issue in light of the large disease spectrum of TB. TB manifestations can range from latency or subclinical disease [57] to increased severity [17]. To bring clarity to these topics, some authors are addressing the host and pathogen simultaneously. For instance, a significant association was reported between a nonsynonymous substitution (G254K) on the Arachidonate 5- Lipoxygenase (ALOX5) gene and increased predisposition to develop TB due to lineage 6. Accordingly, the variant G254K of ALOX5 is highly frequent in Africa when compared to other world regions [58], indicating that lineage 6 strains (abundant in West Africa) might have a selective advantage in hosts harboring the referred polymorphism. In a genome-wide association study (GWAS) with nearly 700 M. tuberculosis clinical isolates, a SNP near the leukocyte surface antigen CD53 gene was significantly associated with older age onset TB cases caused by lineages other than lineage 2 [59]. Interestingly, results are in agreement with the different transmission dynamics by lineage described for Ho Chi Minh City (Vietnam) [54]. On the other hand, recent work [17] could not find a single SNP explaining all the association between severe TB and M. tuberculosis clinical isolates inducing low levels of IL-1β secretion. Instead, several candidate mutations were found, with possible impacts at the protein level (ESX-1 secretion system) and transcription regulation (sigma factor sigA). These data show a complex interaction between the bacterium and the immune system of its host.
CD4+ T cells are crucial immune components to control M. tuberculosis [8]. This is particularly evident when viewed in the context of HIV-1 co-infection. A molecular epidemiology study performed by Lukas Fenner and colleagues [7] showed a significantly higher percentage of active TB infections due to allopatric M. tuberculosis strains (i.e., belonging to lineages not usually associated with the patient’s country of origin) in cases of HIV-1 co-infection. The statistical association was stronger in patients with lower CD4+ T cell counts and remained significant after adjusting for sex, age, contact with foreign populations and immune comorbidities. Thus, these results highlight the importance of studying the immune synapse between bacterial peptides and different human leukocyte antigen (HLA) class II proteins. However, a large majority of the experimentally validated T cell epitopes were described to be hyperconserved across the MTBC [60][61]. Nonetheless, amino acid mutations in predicted CD4+ T cell epitopes were identified in a comprehensive immunoinformatic screening aiming at highly diverse genes. Furthermore, amino acid variants in 10 out of 14 experimentally tested epitopes led to differential immune responses, as measured by IFN-y secretion in the blood of 82 individuals from the Gambia, latently infected with M. tuberculosis [61]. Other elaborated approaches have been also uncovering relevant CD4+ T cell epitopes, including some that are highly prevalent in certain MTBC lineages [62][63][64]. Another important feature of the immune response in TB is the balance between pathogen clearance and immunopathology [8][10]. In this context, a recent study reported the death of a patient with Merkel cell carcinoma after the use of anti-T cell inhibitory receptor PD-1 as an immunotherapeutic strategy, possibly caused by an exacerbated immune response [65]. Overall, immune findings highlight the need for an integrative approach to diagnose and treat TB, taking into account important comorbidities such as the ones caused by HIV-1.
References
- WHO. World Health Organization Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2020.
- WHO. WHO Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016; World Health Organization: Geneva, Switzerland, 2018.
- Joint United Nations Programme on HIV/AIDS UNAIDS Data 2020; UNAIDS: Geneva, Switzerland, 2020.
- Alemu, Y.M.; Awoke, W.; Wilder-Smith, A. Determinants for tuberculosis in HIV-infected adults in Northwest Ethiopia: A multicentre case–control study. BMJ Open 2016, 6, e009058, doi:10.1136/bmjopen-2015-009058.
- Pawlowski, A.; Jansson, M.; Sköld, M.; Rottenberg, M.E.; Källenius, G. Tuberculosis and HIV Co-Infection. PLoS Pathog. 2012, 8, e1002464, doi:10.1371/journal.ppat.1002464.
- Schutz, C.; Meintjes, G.; Almajid, F.; Wilkinson, R.J.; Pozniak, A. Clinical management of tuberculosis and HIV-1 co-infection. Respir. J. 2010, 36, 1460–1481, doi:10.1183/09031936.00110210.
- Fenner, L.; Egger, M.; Bodmer, T.; Furrer, H.; Ballif, M.; Battegay, M.; Helbling, P.; Fehr, J.; Gsponer, T.; Rieder, H.L.; et al. HIV infection disrupts the sympatric host-pathogen relationship in human tuberculosis. PLoS Genet 2013, 9, e1003318, doi:10.1371/journal.pgen.1003318.
- Cooper, A.M. Cell-mediated immune responses in tuberculosis. Rev. Immunol. 2009, 27, 393–422, doi:10.1146/annurev.immunol.021908.132703.
- Dookie, N.; Rambaran, S.; Padayatchi, N.; Mahomed, S.; Naidoo, K. Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. Antimicrob. Chemother. 2018, 73, 1138–1151, doi:10.1093/jac/dkx506.
- Bastos, H.N.; Osório, N.S.; Gagneux, S.; Comas, I.; Saraiva, M. The Troika Host–Pathogen–Extrinsic Factors in Tuberculosis: Modulating Inflammation and Clinical Outcomes. Immunol. 2018, 8, 1948, doi:10.3389/fimmu.2017.01948.
- Galagan, J.E. Genomic insights into tuberculosis. Rev. Genet. 2014, 15, 307–320, doi:10.1038/nrg3664.
- Menardo, F.; Duchêne, S.; Brites, D.; Gagneux, S. The molecular clock of Mycobacterium tuberculosis. PLOS Pathog. 2019, 15, e1008067, doi:10.1371/journal.ppat.1008067.
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Genet. 2013, 45, 1176–1182, doi:10.1038/ng.2744.
- Warner, D.F.; Koch, A.; Mizrahi, V. Diversity and disease pathogenesis in Mycobacterium tuberculosis. Trends Microbiol. 2015, 23, 14–21, doi:10.1016/j.tim.2014.10.005.
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Immunol. 2014, 26, 431–444, doi:10.1016/j.smim.2014.09.012.
- Moreira-Teixeira, L.; Tabone, O.; Graham, C.M.; Singhania, A.; Stavropoulos, E.; Redford, P.S.; Chakravarty, P.; Priestnall, S.L.; Suarez-Bonnet, A.; Herbert, E.; et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Immunol. 2020, 21, 464–476, doi:10.1038/s41590-020-0610-z.
- Sousa, J.; Cá, B.; Maceiras, A.R.; Simões-Costa, L.; Fonseca, K.L.; Fernandes, A.I.; Ramos, A.; Carvalho, T.; Barros, L.; Magalhães, C.; et al. Mycobacterium tuberculosis associated with severe tuberculosis evades cytosolic surveillance systems and modulates IL-1β production. Commun. 2020, 11, 1–14, doi:10.1038/s41467-020-15832-6.
- Gagneux, S.; DeRiemer, K.; Van, T.; Kato-Maeda, M.; de Jong, B.C.; Narayanan, S.; Nicol, M.; Niemann, S.; Kremer, K.; Gutierrez, M.C.; et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Natl. Acad. Sci. USA 2006, 103, 2869–2873, doi:10.1073/pnas.0511240103.
- Yimer, S.A.; Norheim, G.; Namouchi, A.; Zegeye, E.D.; Kinander, W.; Tønjum, T.; Bekele, S.; Mannsåker, T.; Bjune, G.; Aseffa, A.; et al. Mycobacterium tuberculosis Lineage 7 Strains Are Associated with Prolonged Patient Delay in Seeking Treatment for Pulmonary Tuberculosis in Amhara Region, Ethiopia. Clin. Microbiol. 2015, 53, 1301–1309, doi:10.1128/JCM.03566-14.
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Rev. 2015, 264, 6–24, doi:10.1111/imr.12264.
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Rev. Microbiol. 2018, 16, 202–213, doi:10.1038/nrmicro.2018.8.
- O’Neill, M.B.; Shockey, A.; Zarley, A.; Aylward, W.; Eldholm, V.; Kitchen, A.; Pepperell, C.S. Lineage specific histories of Mycobacterium tuberculosis dispersal in Africa and Eurasia. Ecol. 2019, 28, 3241–3256, doi:10.1111/mec.15120.
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Liu, Q.; Trauner, A.; Fenner, L.; Rutaihwa, L.; Borrell, S.; Luo, T.; et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Genet. 2016, 48, 1535–1543, doi:10.1038/ng.3704.
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271, doi:10.1016/S0140-6736(14)60164-1.
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. HIV Sequence Compendium 2018; Theoretical Biology and Biophysics Group, Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2018. .
- Santos, A.F.; Soares, M.A. HIV Genetic Diversity and Drug Resistance. Viruses 2010, 2, 503–531, doi:10.3390/v2020503.
- Beloukas, A.; Psarris, A.; Giannelou, P.; Kostaki, E.; Hatzakis, A.; Paraskevis, D. Molecular epidemiology of HIV-1 infection in Europe: An overview. Genet. Evol. 2016, 46, 180–189, doi:10.1016/j.meegid.2016.06.033.
- Kiwanuka, N.; Laeyendecker, O.; Robb, M.; Kigozi, G.; Arroyo, M.; McCutchan, F.; Eller, L.A.; Eller, M.; Makumbi, F.; Birx, D.; et al. Effect of Human Immunodeficiency Virus Type 1 (HIV-1) Subtype on Disease Progression in Persons from Rakai, Uganda, with Incident HIV-1 Infection. Infect. Dis. 2008, 197, 707–713, doi:10.1086/527416.
- Désiré, N.; Cerutti, L.; Le Hingrat, Q.; Perrier, M.; Emler, S.; Calvez, V.; Descamps, D.; Marcelin, A.G.; Hué, S.; Visseaux, B. Characterization update of HIV-1 M subtypes diversity and proposal for subtypes A and D sub-subtypes reclassification. Retrovirology 2018, 15, 80, doi:10.1186/s12977-018-0461-y.
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. ISRN Microbiol. 2013, 2013, 1–20, doi:10.1155/2013/481314.
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.B.; Warren, R.; Dippenaar, A.; Parsons, S.D.C.; Beisel, C.; Behr, M.A.; et al. A New Phylogenetic Framework for the Animal-Adapted Mycobacterium tuberculosis Complex. Microbiol. 2018, 9, 2820, doi:10.3389/fmicb.2018.02820.
- Chiner-Oms, Á.; Sánchez-Busó, L.; Corander, J.; Gagneux, S.; Harris, S.R.; Young, D.; González-Candelas, F.; Comas, I. Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex. Adv. 2019, 5, eaaw3307, doi:10.1126/sciadv.aaw3307.
- Riojas, M.A.; McGough, K.J.; Rider-Riojas, C.J.; Rastogi, N.; Hazbón, M.H. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycob. J. Syst. Evol. Microbiol. 2018, 68, 324–332, doi:10.1099/ijsem.0.002507.
- Duffy, S.C.; Srinivasan, S.; Schilling, M.A.; Stuber, T.; Danchuk, S.N.; Michael, J.S.; Venkatesan, M.; Bansal, N.; Maan, S.; Jindal, N.; et al. Reconsidering Mycobacterium bovis as a proxy for zoonotic tuberculosis: A molecular epidemiological surveillance study. Lancet Microbe 2020, 1, e66–e73, doi:10.1016/s2666-5247(20)30038-0.
- Ghodbane, R.; Drancourt, M. Non-human sources of Mycobacterium tuberculosis. Tuberculosis 2013, 93, 589–595, doi:10.1016/J.TUBE.2013.09.005.
- Villarreal-Ramos, B.; Berg, S.; Whelan, A.; Holbert, S.; Carreras, F.; Salguero, F.J.; Khatri, B.L.; Malone, K.; Rue-Albrecht, K.; Shaughnessy, R.; et al. Experimental infection of cattle with Mycobacterium tuberculosis isolates shows the attenuation of the human tubercle bacillus for cattle. Rep. 2018, 8, 894, doi:10.1038/s41598-017-18575-5.
- Ngabonziza, J.C.S.; Loiseau, C.; Marceau, M.; Jouet, A.; Menardo, F.; Tzfadia, O.; Antoine, R.; Niyigena, E.B.; Mulders, W.; Fissette, K.; et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Commun. 2020, 11, 1–11, doi:10.1038/s41467-020-16626-6.
- Coscolla, M.; Brites, D.; Menardo, F.; Loiseau, C.; Borrell, S.; Otchere, I.D.; Asante-Poku, A.; Asare, P.; Sánchez-Busó, L.; Gehre, F.; et al. Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history. bioRxiv 2020, doi:10.1101/2020.06.10.141788.
- Sabin, S.; Herbig, A.; Vågene, Å.J.; Ahlström, T.; Bozovic, G.; Arcini, C.; Kühnert, D.; Bos, K.I. A seventeenth-century Mycobacterium tuberculosis genome supports a Neolithic emergence of the Mycobacterium tuberculosis complex. Genome Biol. 2020, 21, 201, doi:10.1186/s13059-020-02112-1.
- Eldholm, V.; Pettersson, J.H.-O.; Brynildsrud, O.B.; Kitchen, A.; Rasmussen, E.M.; Lillebaek, T.; Rønning, J.O.; Crudu, V.; Mengshoel, A.T.; Debech, N.; et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Natl. Acad. Sci. USA 2016, 113, 201611283, doi:10.1073/pnas.1611283113.
- Brynildsrud, O.B.; Pepperell, C.S.; Suffys, P.; Grandjean, L.; Monteserin, J.; Debech, N.; Bohlin, J.; Alfsnes, K.; Pettersson, J.O.-H.; Kirkeleite, I.; et al. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Adv. 2018, 4, eaat5869, doi:10.1126/sciadv.aat5869.
- de Jong, B.C.; Hill, P.C.; Aiken, A.; Awine, T.; Antonio, M.; Adetifa, I.M.; Jackson-Sillah, D.J.; Fox, A.; DeRiemer, K.; Gagneux, S.; et al. Progression to Active Tuberculosis, but Not Transmission, Varies by Mycobacterium tuberculosis Lineage in The Gambia. Infect. Dis. 2008, 198, 1037–1043, doi:10.1086/591504.
- Tientcheu, L.D.; Koch, A.; Ndengane, M.; Andoseh, G.; Kampmann, B.; Wilkinson, R.J. Immunological consequences of strain variation within the Mycobacterium Tuberculosis complex. J. Immunol. 2017, 47, 432–445, doi:10.1002/eji.201646562.
- Harishankar, M.; Selvaraj, P.; Bethunaickan, R. Influence of Genetic Polymorphism Towards Pulmonary Tuberculosis Susceptibility. Med. 2018, 5, 213, doi:10.3389/fmed.2018.00213.
- Wiens, K.E.; Woyczynski, L.P.; Ledesma, J.R.; Ross, J.M.; Zenteno-Cuevas, R.; Goodridge, A.; Ullah, I.; Mathema, B.; Djoba Siawaya, J.F.; Biehl, M.H.; et al. Global variation in bacterial strains that cause tuberculosis disease: A systematic review and meta-analysis. BMC Med. 2018, 16, 196, doi:10.1186/s12916-018-1180-x.
- Comas, I.; Hailu, E.; Kiros, T.; Bekele, S.; Mekonnen, W.; Gumi, B.; Tschopp, R.; Ameni, G.; Hewinson, R.G.; Robertson, B.D.; et al. Population Genomics of Mycobacterium tuberculosis in Ethiopia Contradicts the Virgin Soil Hypothesis for Human Tuberculosis in Sub-Saharan Africa. Biol. 2015, 25, 3260–3266, doi:10.1016/j.cub.2015.10.061.
- Gagneux, S. Host-pathogen coevolution in human tuberculosis. Trans. R. Soc. Lond. B. Biol. Sci. 2012, 367, 850–859, doi:10.1098/rstb.2011.0316.
- Palittapongarnpim, P.; Ajawatanawong, P.; Viratyosin, W.; Smittipat, N.; Disratthakit, A.; Mahasirimongkol, S.; Yanai, H.; Yamada, N.; Nedsuwan, S.; Imasanguan, W.; et al. Evidence for Host-Bacterial Co-evolution via Genome Sequence Analysis of 480 Thai Mycobacterium tuberculosis Lineage 1 Isolates. Rep. 2018, 8, 11597, doi:10.1038/s41598-018-29986-3.
- Couvin, D.; David, A.; Zozio, T.; Rastogi, N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Genet. Evol. 2019, 72, 31–43, doi:10.1016/j.meegid.2018.12.030.
- Tulu, B.; Ameni, G. Spoligotyping based genetic diversity of Mycobacterium tuberculosis in Ethiopia: A systematic review. BMC Infect. Dis. 2018, 18, 140, doi:10.1186/s12879-018-3046-4.
- Jacobs, W.R.; McShane, H.; Mizrahi, V.; Orme, I.M. Tuberculosis and the Tubercle Bacillus; ASM Books; Wiley: Hoboken, NJ, USA, 2020; ISBN 9781555819569.
- Freschi, L.; Vargas Jr, R.; Hussain, A.; Mostofa Kamal, S.M.; Skrahina, A.; Tahseen, S.; Ismail, N.; Barbova, A.; Niemann, S.; Maria Cirillo, D.; et al. Population structure, biogeography and transmissibility of Mycobacterium tuberculosis. bioRxiv 2020, doi:10.1101/2020.09.29.293274.
- Cowley, D.; Govender, D.; February, B.; Wolfe, M.; Steyn, L.; Evans, J.; Wilkinson, R.J.; Nicol, M.P. Recent and rapid emergence of W-Beijing strains of Mycobacterium tuberculosis in Cape Town, South Africa. Infect. Dis. 2008, 47, 1252–1259, doi:10.1086/592575.
- Holt, K.E.; McAdam, P.; Thai, P.V.K.; Thuong, N.T.T.; Ha, D.T.M.; Lan, N.N.; Lan, N.H.; Nhu, N.T.Q.; Hai, H.T.; Ha, V.T.N.; et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Genet. 2018, 50, 849–856, doi:10.1038/s41588-018-0117-9.
- Rutaihwa, L.K.; Menardo, F.; Stucki, D.; Gygli, S.M.; Ley, S.D.; Malla, B.; Feldmann, J.; Borrell, S.; Beisel, C.; Middelkoop, K.; et al. Multiple Introductions of Mycobacterium tuberculosis Lineage 2–Beijing Into Africa Over Centuries. Ecol. Evol. 2019, 7, 112, doi:10.3389/fevo.2019.00112.
- Ford, C.B.; Shah, R.R.; Maeda, M.K.; Gagneux, S.; Murray, M.B.; Cohen, T.; Johnston, J.C.; Gardy, J.; Lipsitch, M.; Fortune, S.M. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Genet. 2013, 45, 784–790, doi:10.1038/ng.2656.
- Xu, Y.; Cancino-Muñoz, I.; Torres-Puente, M.; Villamayor, L.M.; Borrás, R.; Borrás-Máñez, M.; Bosque, M.; Camarena, J.J.; Colomer-Roig, E.; Colomina, J.; et al. High-resolution mapping of tuberculosis transmission: Whole genome sequencing and phylogenetic modelling of a cohort from Valencia Region, Spain. PLOS Med. 2019, 16, e1002961, doi:10.1371/journal.pmed.1002961.
- Herb, F.; Thye, T.; Niemann, S.; Browne, E.N.L.; Chinbuah, M. a.; Gyapong, J.; Osei, I.; Owusu-Dabo, E.; Werz, O.; Rüsch-Gerdes, S.; et al. ALOX5 variants associated with susceptibility to human pulmonary tuberculosis. Mol. Genet. 2008, 17, 1052–1060, doi:10.1093/hmg/ddm378.
- Omae, Y.; Toyo-oka, L.; Yanai, H.; Nedsuwan, S.; Wattanapokayakit, S.; Satproedprai, N.; Smittipat, N.; Palittapongarnpim, P.; Sawanpanyalert, P.; Inunchot, W.; et al. Pathogen lineage-based genome-wide association study identified CD53 as susceptible locus in tuberculosis. Hum. Genet. 2017, 62, 1015–1022, doi:10.1038/jhg.2017.82.
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Genet. 2010, 42, 498–503, doi:10.1038/ng.590.
- Coscolla, M.; Copin, R.; Sutherland, J.; Gehre, F.; de Jong, B.; Owolabi, O.; Mbayo, G.; Giardina, F.; Ernst, J.D.; Gagneux, S.M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe 2015, 18, 538–548, doi:10.1016/j.chom.2015.10.008.
- Coppola, M.; Ottenhoff, T.H. Genome wide approaches discover novel Mycobacterium tuberculosis antigens as correlates of infection, disease, immunity and targets for vaccination. Immunol. 2018, 39, 88–101, doi:10.1016/J.SMIM.2018.07.001.
- Otchere, I.D.; Coscollá, M.; Sánchez-Busó, L.; Asante-Poku, A.; Brites, D.; Loiseau, C.; Meehan, C.; Osei-Wusu, S.; Forson, A.; Laryea, C.; et al. Comparative genomics of Mycobacterium africanum Lineage 5 and Lineage 6 from Ghana suggests distinct ecological niches. Rep. 2018, 8, 11269, doi:10.1038/s41598-018-29620-2.
- Vyakarnam, A.; Ramaiah, A.; Nayak, S.; Rakshit, S.; Manson, A.L.; Abeel, T.; Shanmugam, S.; Sahoo, P.N.; UKJ, A.J.; Sundaramurthi, J.C.; et al. Evidence for Highly Variable, Region-Specific Patterns of T-cell Epitope Mutations Accumulating in Mycobacterium tuberculosis Strains. Immunol. 2019, 10, 195, doi:10.3389/FIMMU.2019.00195.
- Barber, D.L.; Sakai, S.; Kudchadkar, R.R.; Fling, S.P.; Day, T.A.; Vergara, J.A.; Ashkin, D.; Cheng, J.H.; Lundgren, L.M.; Raabe, V.N.; et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Transl. Med. 2019, 11, eaat2702, doi:10.1126/scitranslmed.aat2702.

