 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robrecht Raedt | + 2611 word(s) | 2611 | 2021-01-07 04:17:52 | | | |
| 2 | Dean Liu | -857 word(s) | 1754 | 2021-01-19 13:16:00 | | |
Video Upload Options
Adenosine acts as an endogenous anticonvulsant and seizure terminator in the brain. Many of its anticonvulsive effects are mediated through activation of the adenosine A1 receptor, a G protein-coupled receptor with a wide array of targets. Various signaling pathways are involved in the neuronal inhibition caused by adenosine A1 receptor activation. These include direct interactions of G protein subunits, the adenyl cyclase pathway and the phospholipase C pathway, which all mediate neuronal hyperpolarization and suppression of synaptic transmission.
1. Introduction
Epilepsy is a chronic brain disease ranking among the most common neurological disorders with an estimated prevalence of around 1% worldwide[1][2]. First-line treatment consists of pharmacotherapy with anti-epileptic drugs. Despite the development and approval of more than 20 new drugs over the past few decades, about one third of all epilepsy patients cannot be effectively treated this way[3][4]. This significant proportion of patients suffering from drug-resistant epilepsy has been an important drive for the search for new and better epilepsy treatments. In this regard, a lot of research has focused on the role of adenosine in epilepsy, owing to its ability to act as an endogenous seizure terminator and its potent anticonvulsive effects[5][6][7]. A great deal of studies have examined the mechanisms behind the anti-epileptic effects of adenosine and demonstrated that adenosine or adenosine analogues are effective in suppressing epileptic seizures, and this mainly through activation of adenosine A1 receptors. Several excellent reviews have been published in recent years describing the current knowledge on the role of adenosine in epilepsy and its therapeutic potential (see references[8][9][10]).
2. Adenosine in the Central Nervous System
Adenosine is a purine ribonucleoside fulfilling an important role in many physiological processes[11]. It has a general homeostatic role as modulator of cellular metabolism, but in the central nervous system (CNS) it also distinctively functions as a neuromodulator[12]. Adenosine is involved in various neural processes, including the regulation of sleep, arousal, nociception and respiration[13][14][15][16].
Adenosine is constitutively present at low concentrations in the brain, with basal extracellular adenosine levels kept in the range of 50–200 nM through enzymatic control[17]. The main source of adenosine in the brain is the intra- and extracellular breakdown of adenine nucleotides by 5′-nucleotidases (Figure 1). Adenine nucleotides released in the extracellular space, such as adenosine triphosphate (ATP) or adenosine monophosphate (AMP), are rapidly converted to adenosine[18]. Intracellularly, the formation of adenosine is linked to the energy consumption of the cell. An increase in cellular workload and in degradation of cytoplasmic ATP leads to increased formation of adenosine, with small intracellular changes in the concentration of ATP resulting in substantial changes in adenosine concentrations relative to its basal levels[12][19]. Adenosine formed intracellularly then exits the cell via equilibrative nucleoside transporters (ENTs), which allow for bidirectional passive transport of adenosine according to the concentration gradient. This way, extracellular adenosine concentration is mainly regulated via two intracellular enzymes: adenosine deaminase (ADA), which catabolizes adenosine to inosine, and adenosine kinase (ADK), which phosphorylates it to AMP[17][20]. Under physiological conditions, ADK is the main regulator of adenosine concentrations, but when concentrations increase in case of energy imbalance ADA exerts a more important role [21].

Figure 1. Adenosine metabolism in the brain: intra-(IC) and extracellular (EC) catabolization of adenine nucleotides (ATP, ADP, AMP) by nucleotidases (NT) leads to formation of adenosine. Intracellularly, adenosine deaminase (ADA) breaks down adenosine to inosine and adenosine kinase (ADK) phosphorylates adenosine to AMP. Bidirectional transport of adenosine via equilibrative nucleoside transporters (ENT) equalizes the IC and EC adenosine concentrations.
Extracellular adenosine exerts its modulatory effects via binding to G protein-coupled receptors (GPCRs), of which four subtypes have been characterized: A1, A2A, A2B and A3. These subtypes possess different affinities for adenosine and couple to specific G proteins. The adenosine A1 receptor (A1R) couples to Gi and Go proteins, the adenosine A2A receptor (A2AR) couples to Gs and Golf proteins, the adenosine A2B receptor (A2BR) couples to Gs and Gq proteins and the adenosine A3 receptor (A3R) couples to Gi and Gq proteins[20]. The A1 and A2A subtypes are high affinity receptors, with the A1R possessing the highest affinity for adenosine. They are the most abundantly expressed adenosine receptors in the CNS, while the A2BR and A3R have much lower affinities and are only expressed there in comparatively small numbers[13]. Highest CNS expression levels of the A1R are found in the neocortex, hippocampus, thalamus, cerebellum and spinal cord. The A2ARs on the other hand are predominantly expressed in the striatum[22].
3. A1R Structure, Activation and Expression
The A1R, together with the other adenosine receptors, belongs to the GPCR superfamily and is further classified into the α subfamily of the rhodopsins (formerly called “class A” of the GPCR superfamily)[23]. It is a glycoprotein with a molecular mass of ~36 kDa and, like all GPCRs, consists of 7 transmembrane α-helices, 3 extracellular and 3 intracellular loops, an extracellular N-terminus and an intracellular C-terminus[23][24]. The first four transmembrane domains of the A1R, (from the N-terminus to the end of the second extracellular loop) have been shown to be important for ligand binding and conferring specificity for A1-selective agonists/antagonists[25]. More recently, the determination of the crystal structure of the A1R in its inactive state has confirmed that conformational differences in these regions, especially the distinct conformation of the second extracellular loop, could underlie the selectivity of ligands for the A1 subtype[26]. Binding of an agonist to the A1R induces structural changes leading to receptor activation. The overall activation process is similar for all GPCRs and involves the relative rearrangement of transmembrane helices. A key transition during activation is the outward movement of the intracellular part of the transmembrane helix 6 (Figure 2), which has been observed in multiple GPCRs including the adenosine-bound A1R [27]. This opens up the cytosolic side of the receptor and enables interaction with G proteins, resulting in a ternary complex between agonist, receptor and G protein. Experiments with fusion proteins of the A1R and G protein subunits have indicated that receptor activation is the rate-limiting step in this ternary complex formation, rather than the interaction between the activated receptor and the G protein[28]. The kinetics of this activation process have been studied by looking at conformational changes with fluorescence resonance energy transfer (FRET) sensors. In these studies, receptor activation times were indirectly measured in various GPCRs and were found to be in the range of 30–50 ms[29].
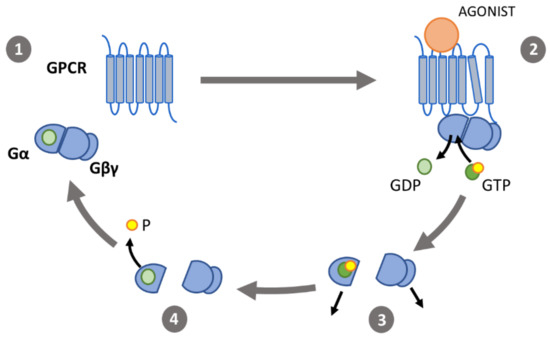
Figure 2. The G protein activation cycle: (1) in its inactive state, the α-subunit (Gα) binds guanosine diphosphate (GDP) and forms a heterotrimeric G protein complex with the β- and γ-subunits (Gβγ). (2) Binding of an agonist to a G-protein coupled receptor (GPCR) induces conformational changes. The outward movement of transmembrane helix 6 enables interaction of the GPCR with the heterotrimeric G proteins, catalyzing the exchange of GDP for GTP. (3) Gα and Gβγ then dissociate and interact with effectors. (4) Gα-induced hydrolyzation of GTP to GDP causes the G protein subunits to associate and return to their inactive state.
The gene coding for the human A1R is located on the long arm of chromosome 1 and contains two separate promotors; A and B[30][31]. This results in two distinct transcripts of the A1R gene: transcript α produced by promoter A and transcript β produced by promoter B. Transcript β is found in all tissues expressing A1Rs while transcript α is only seen in tissues with high levels of A1R expression, such as the brain, testis and kidney[31]. This is due to multiple AUG codons in the 5′-untranslated region of transcript β which hinder protein expression at the post-transcriptional level[32]. In the CNS, A1Rs are most abundant in neurons, but A1Rs are also expressed by astrocytes, microglia and oligodendrocytes[33]. Receptor distribution varies per region, with the highest densities of A1Rs being found in the hippocampus[34]. The subcellular localization has been investigated in rat hippocampal neurons, where A1Rs are present extrasynaptically on the membrane of cell bodies, axons and dendrites and synaptically in the active zone of presynaptic terminals and at the postsynaptic density[35][36][37][38].
4. A1R signaling pathways leading to neuronal inhibition
The two principal inhibitory neuronal mechanisms of the A1R are well known; (1) membrane hyperpolarization caused by the activation of K+ channels and (2) suppression of synaptic transmission via inhibition of VGCCs and synaptic vesicle release (Figure 3). The second messenger systems and molecular mechanisms responsible for the activation or inhibition of these targets, however, remain to be completely unraveled, though evidence indicates important roles of the AC and the PLC pathways, along with the Gβγ subunit (Figure 4, Figure 5).
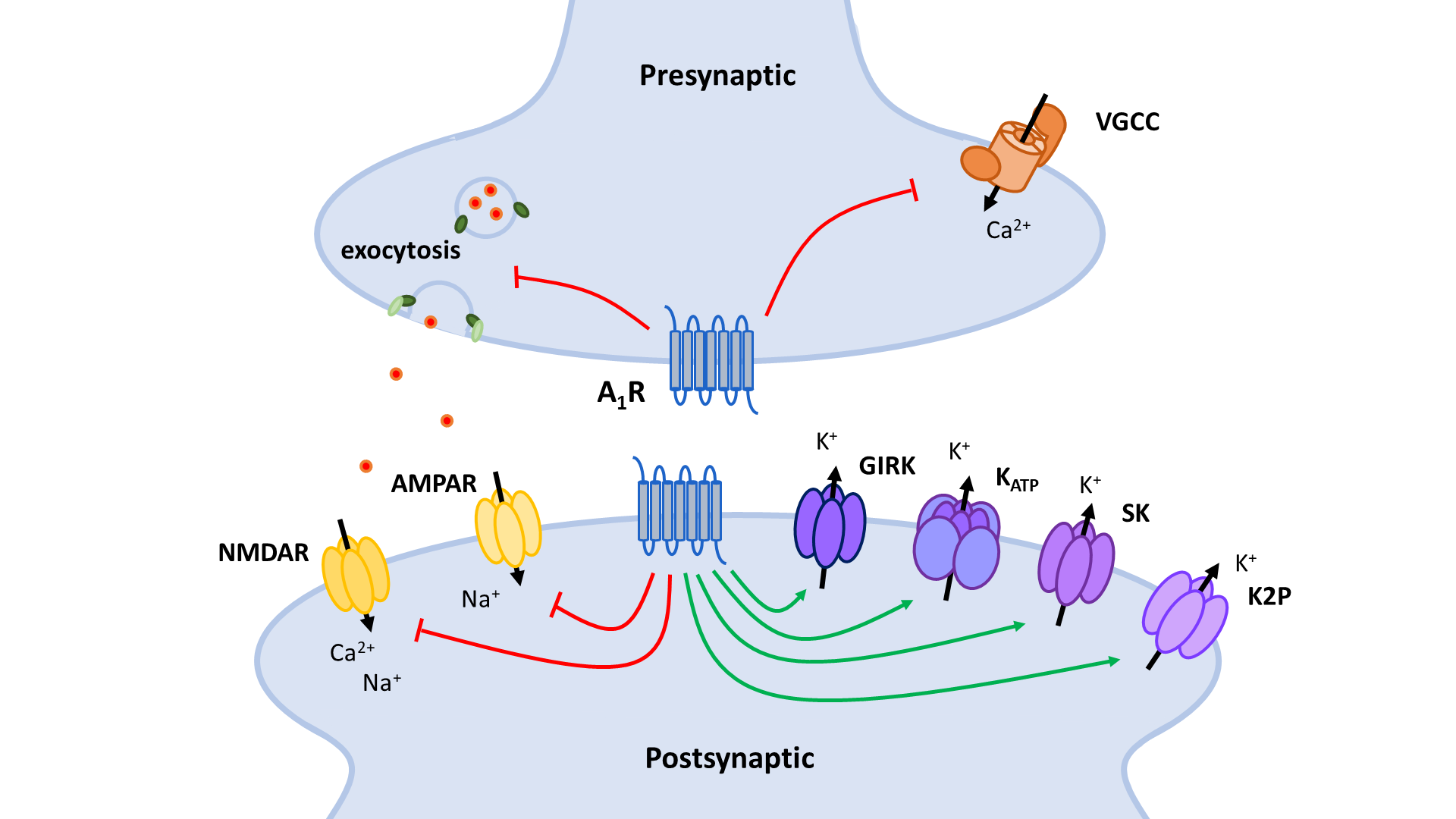
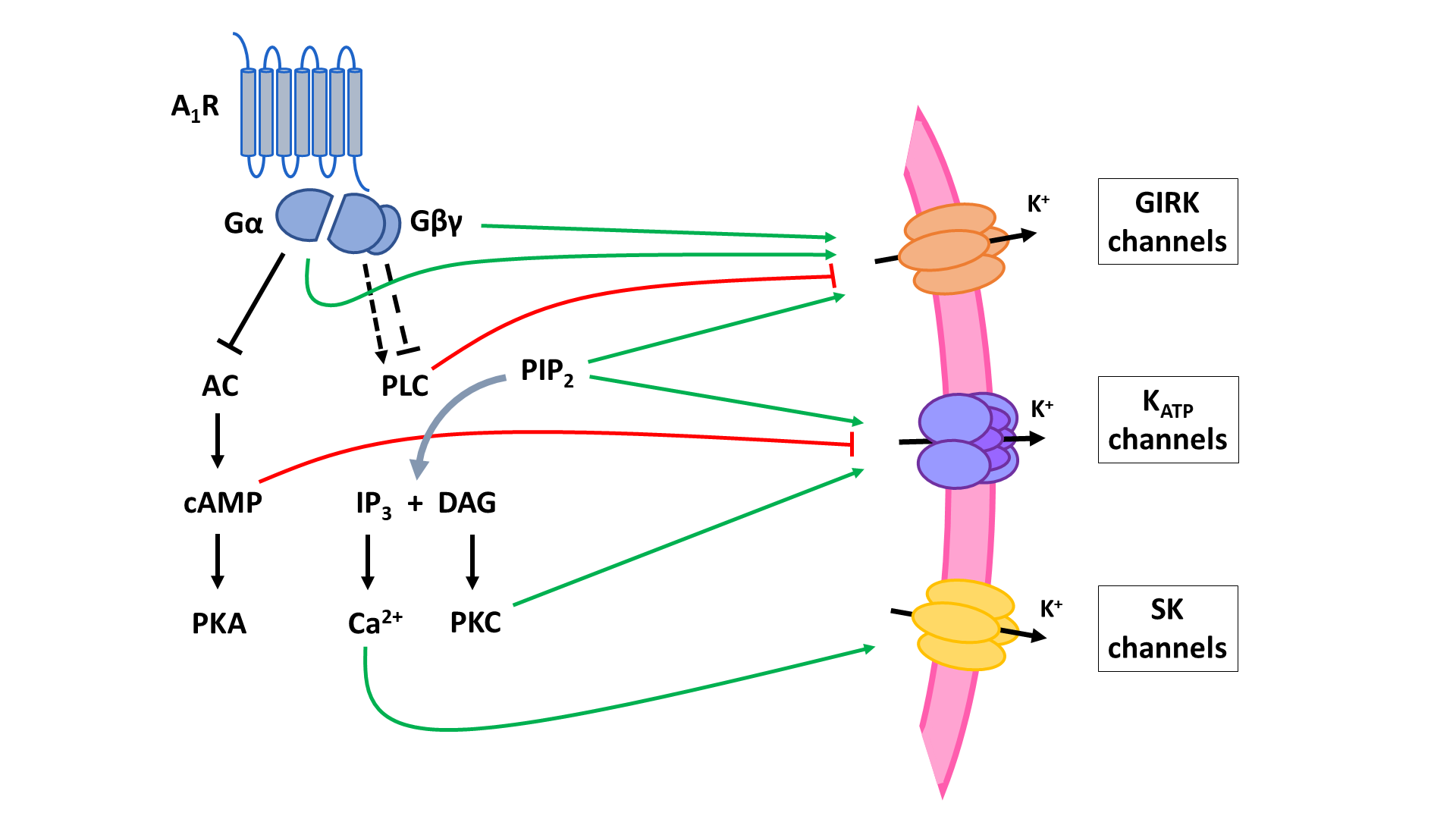
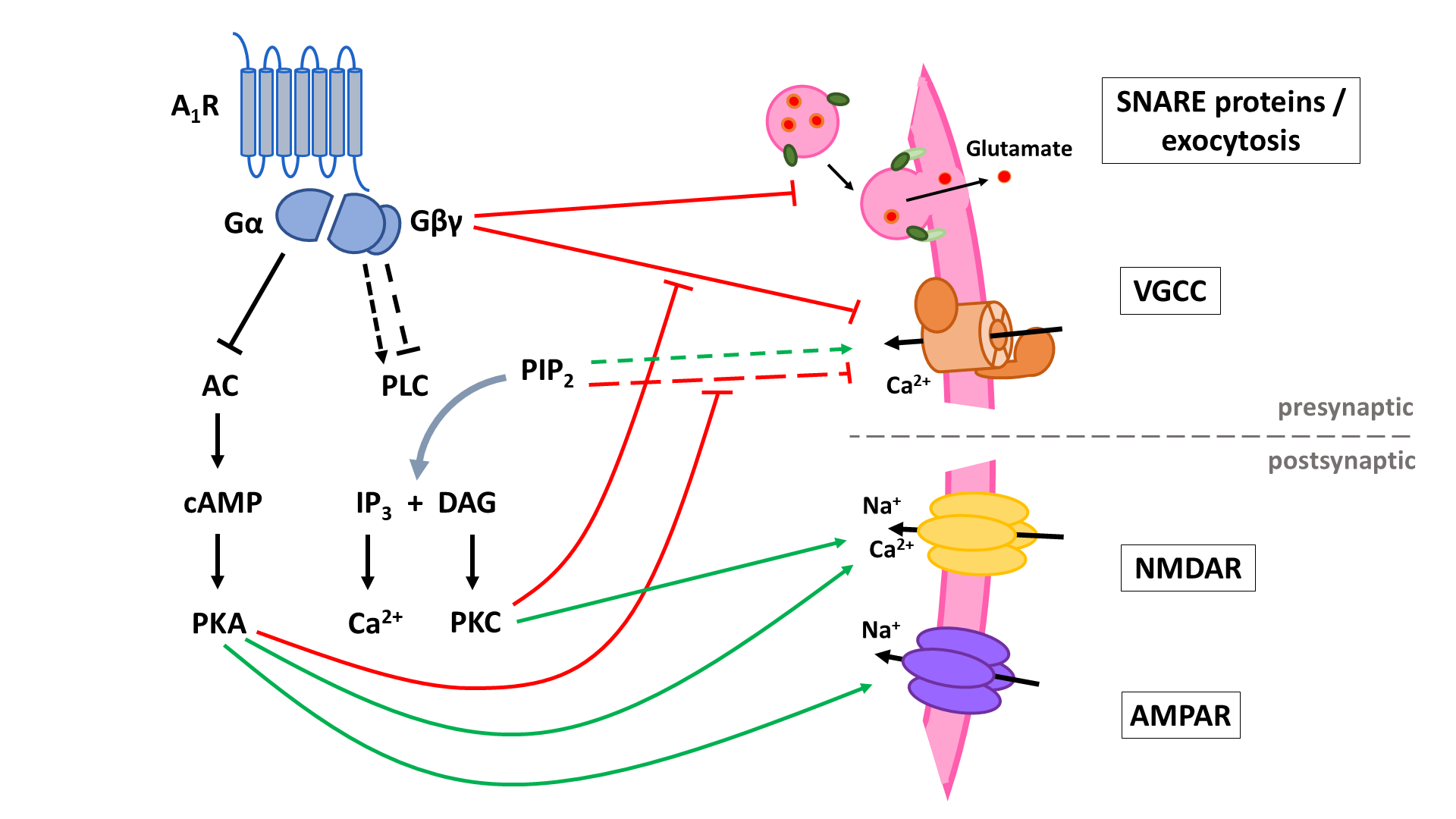
A1 receptors (A1R). A1Rs suppress neurotransmitter release in a Ca2+-dependent way by inhibiting voltage-gated Ca2+ channels (VGCCs) via Gβγ. Additionally, VGCCs are inhibited through reduced PLC signaling resulting in reduced disinhibition by PKC and increased inhibition by PIP2. Inhibition of PKA activity by A1R also enhances PIP2-mediated inhibition of VGCCs. Through binding of Gβγ to SNARE proteins, A1Rs also suppress neurotransmitter release in a Ca2+-independent way. Postsynaptic NMDA (NMDAR) and AMPA receptor (AMPAR) function is negatively modulated by A1Rs through inhibition of PKA and PKC activity. AC: adenyl cyclase; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; PLC: phospholipase C; PIP2: phosphatidylinositol 4,5-bisphosphate; IP3: inositol 1,4,5-trisphosphate; DAG: diacylglycerol; PKC: phosphokinase C.
References
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the Burden of Active and Life-Time Epilepsy: A Meta-Analytic Approach. Epilepsia 2010, 51, 883–890.
- Behr, C.; Goltzene, M.A.; Kosmalski, G.; Hirsch, E.; Ryvlin, P. Epidemiology of Epilepsy. Rev. Neurol. (Paris) 2016, 172, 27–36.
- Golyala, A.; Kwan, P. Drug Develpment for Refractory Epilepsy: The Past 25 Years and Beyond. Seizure 2017, 44, 147–156.
- Kwan, P.; Brodie, M.J. Early Identification of Refractory Epilepsy. N. Engl. J. Med. 2000, 342, 314–319.
- Dunwiddie, T.V. Endogenously Released Adenosine Regulates Excitability in the In Vitro Hippocampus. Epilepsia 1980, 21, 541–548.
- Dragunow, M.; Goddard, G.V.; Laverty, R. Is Adenosine an Endogenous Anticonvulsant? Epilepsia 1985, 26, 480–487.
- During, M.J.; Spencer, D.D. Adenosine: A Potential Mediator of Seizure Arrest and Postictal Refractoriness. Ann. Neurol. 1992, 32, 618–624.
- Boison, D. Adenosinergic Signaling in Epilepsy. Neuropharmacology 2016, 104, 131–139.
- Rombo, D.M.; Ribeiro, J.A.; Sebastião, A.M. Role of Adenosine Receptors in Epileptic Seizures. In The Adenosine Receptors; Springer International Publishing: Cham, Switzerland, 2018; Volume 34, pp. 309–350.
- Weltha, L.; Reemmer, J.; Boison, D. The Role of Adenosine in Epilepsy. Brain Res. Bull. 2019, 151, 46–54.
- Sachdeva, S.; Gupta, M. Adenosine and Its Receptors as Therapeutic Targets: An Overview. Saudi Pharm. J. 2013, 21, 245–253.
- Cunha, R.A. Adenosine as a Neuromodulator and as a Homeostatic Regulator in the Nervous System: Different Roles, Different Sources and Different Receptors. Neurochem. Int. 2001, 38, 107–125.
- Dunwiddie, T.V.; Masino, S.A. The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 2001, 24, 31–55.
- Franziska Reichert, C.; Cajochen, C.; Schmidt, C.; Cajochen, C. Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine. Biology (Basel) 2016, 5, 11.
- Sawynok, J.; Liu, X.J. Adenosine in the Spinal Cord and Periphery: Release and Regulation of Pain. Prog. Neurobiol. 2003, 69, 313–340.
- Lahiri, S.; Mitchell, C.H.; Reigada, D.; Roy, A.; Cherniack, N.S. Purines, the Carotid Body and Respiration. Respir. Physiol. Neurobiol. 2007, 157, 123–129.
- Latini, S.; Pedata, F. Adenosine in the Central Nervous System: Release Mechanisms and Extracellular Concentrations. J. Neurochem. 2001, 79, 463–484.
- Dunwiddie, T.V.; Dlao, L.; Proctor, W.R. Adenine Nucleotides Undergo Rapid, Quantitative Conversion to Adenosine in the Extracellular Space in Rat Hippocampus. J. Neurosci. 1997, 17, 7673–7682.
- Fredholm, B.B. Adenosine, an Endogenous Distress Signal, Modulates Tissue Damage and Repair. Cell Death Differ. 2007, 14, 1315–1323.
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.-N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552.
- Lloyd, H.G.E.; Fredholm, B.B. Involvement of Adenosine Deaminase and Adenosine Kinase in Regulating Extracellular Adenosine Concentration in Rat Hippocampal Slices. Neurochem. Int. 1995, 26, 387–395.
- Benarroch, E.E. Adenosine and Its Receptors: Multiple Modulatory Functions and Potential Therapeutic Targets for Neurologic Disease. Neurology 2008, 70, 231–236.
- Latek, D.; Modzelewska, A.; Trzaskowski, B.; Palczewski, K.; Filipek, S. G Protein-Coupled Receptors-Recent Advances. Acta Biochim. Pol. 2012, 59, 515–529.
- Ren, H.; Stiles, G.L. Characterization of the Human A1 Adenosine Receptor Gene. Evidence for Alternative Splicing. J. Biol. Chem. 1994, 269, 3104–3110.
- Rivkees, S.A.; Lasbury, M.E.; Barbhaiya, H. Identification of Domains of the Human A1 Adenosine Receptor That Are Important for Binding Receptor Subtype-Selective Ligands Using Chimeric A1/A2a Adenosine Receptors. J. Biol. Chem. 1995, 270, 20485–20490.
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877.e13.
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the Adenosine-Bound Human Adenosine A1 Receptor-Gi Complex. Nature 2018, 558, 559–565.
- Waldhoer, M.; Wise, A.; Milligan, G.; Freissmuth, M.; Nanoff, C. Kinetics of Ternary Complex Formation with Fusion Proteins Composed of the A1-Adenosine Receptor and G Protein α-Subunits. J. Biol. Chem. 1999, 274, 30571–30579.
- Lohse, M.J.; Maiellaro, I.; Calebiro, D. Kinetics and Mechanism of G Protein-Coupled Receptor Activation. Curr. Opin. Cell Biol. 2014, 27, 87–93.
- Townsend-Nicholson, A.; Baker, E.; Schofield, P.R.; Sutherland, G.R. Localization of the Adenosine A1 Receptor Subtype Gene (ADORA1) to Chromosome 1q32.1. Genomics 1995, 26, 423–425.
- Ren, H.; Stiles, G.L. Separate Promoters in the Human A1 Adenosine Receptor Gene Direct the Synthesis of Distinct Messenger RNAs That Regulate Receptor Abundance. Mol. Pharmacol. 1995, 48, 975–980.
- Ren, H.; Stiles, G.L. Posttranscriptional MRNA Processing as a Mechanism for Regulation of Human A1 Adenosine Receptor Expression. Proc. Natl. Acad. Sci. USA 1994, 91, 4864–4866.
- Cunha, R.A. Neuroprotection by Adenosine in the Brain: From A1 Receptor Activation to A2A Receptor Blockade. Purinergic Signal. 2005, 1, 111–134.
- Svenningsson, P.; Hall, H.; Sedvall, G.; Fredholm, B.B. Distribution of Adenosine Receptors in the Postmortem Human Brain: An Extended Autoradiographic Study. Synapse 1997, 27, 322–335.
- Tetzlaff, W.; Schubert, P.; Kreutzberg, G.W. Synaptic and Extrasynaptic Localization of Adenosine Binding Sites in the Rat Hippocampus. Neuroscience 1987, 21, 869–875.
- Swanson, T.H.; Drazba, J.A.; Rivkees, S.A. Adenosine A1 Receptors Are Located Predominantly on Axons in the Rat Hippocampal Formation. J. Comp. Neurol. 1995, 363, 517–531.
- Ochiishi, T.; Chen, L.; Yukawa, A.; Saitoh, Y.; Sekino, Y.; Arai, T.; Nakata, H.; Miyamoto, H. Cellular Localization of Adenosine A1 Receptors in Rat Forebrain: Immunohistochemical Analysis Using Adenosine A1 Receptor-Specific Monoclonal Antibody. J. Comp. Neurol. 1999, 411, 301–316.
- Rebola, N.; Pinheiro, P.C.; Oliveira, C.R.; Malva, J.O.; Cunha, R.A. Subcellular Localization of Adenosine A1 Receptors in Nerve Terminals and Synapses of the Rat Hippocampus. Brain Res. 2003, 987, 49–58.

