 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jens Hahne | + 3201 word(s) | 3201 | 2021-01-07 09:42:53 | | | |
| 2 | Camila Xu | Meta information modification | 3201 | 2021-01-19 09:21:40 | | | | |
| 3 | Camila Xu | Meta information modification | 3201 | 2021-01-19 09:22:30 | | | | |
| 4 | Camila Xu | Meta information modification | 3201 | 2021-01-19 09:22:56 | | | | |
| 5 | Camila Xu | Meta information modification | 3201 | 2021-01-19 09:23:26 | | |
Video Upload Options
Microsatellite Instability (MSI) is the hallmark of Lynch syndrome and it was first described in colorectal cancer patients in 1913. Later the definition was broadened and extracolonic tumors have been included.
1. Lynch Syndrome and Microsatellite Instability (MSI)
Lynch syndrome patients have either germline mutations in genes coding for DNA mismatch repair genes (like MLH1, MSH2, MSH6, and PMS2) or transcriptional inactivation of these genes. Missing functional DNA mismatch repair proteins results in reduced genome integrity due to missing proof-reading and editing during DNA transcription. Furthermore, variations in microsatellite repetitions occur, thus causing changes in the genome length [1][2][3][4]. This molecular phenotype can be used either as a diagnostic tool by polymerase chain reaction (PCR) amplification of microsatellite sequences or by next-generation sequencing for detection of MSI. Another diagnostic tool is based on immunohistochemistry staining for expression of mismatch repair proteins [2].
In general, Lynch syndrome represents a high risk factor to develop cancer and predisposes to several cancer types including colorectal, endometrial, gastric, ovarian, urinary tract, prostate, small bowel, duodenal, esophageal, hepatocellular, gallbladder, pancreatic cancer, and intrahepatic cholangiocarcinoma [5][6][2][7][8]. Moreover, a defective mismatch repair system results in the accumulation of somatic mutations, leading to higher neo-antigen load, which promotes T-cell activation. Increased neo-antigen expression and recruitment of cytotoxic T-cell can contribute to the increased immunogenicity of cancers with MSI and might enhance the vulnerability of these tumors to immunotherapy [9]. It has been calculated that MSI tumors contain 10 to 100 times more mutations than cancers with an intact DNA mismatch repair system [10].
2. MSI and Pancreatic Cancer
The colorectal tumor is the most common tumor among Lynch syndrome families; however, families that carry the MMR (mismatch repair) gene defect have a very high risk to develop pancreatic cancer. Lynch syndrome patients have a nearly 9-fold higher risk of pancreatic cancer in comparison to the general population. Pancreatic tumors caused by Lynch syndrome have often a medullary appearance with prominent lymphocytic infiltration [5][11][12][13]. Pancreatic cancer developed in Lynch syndrome context is normally diagnosed before the age of 60 [5][14] and some can also have different histological subtypes such as intraductal papillary mucinous neoplasms and acinar cell carcinoma [15][16][17]. MSI has been found in several patients’ studies both on resected and metastatic diseases with frequencies between 0% and 75% (Table 1). This wide difference might be related to the patients’ selection criteria and to the different markers used for mismatch repair detection [18][19][15][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44]. Nevertheless, the overall rate of MSI pancreatic cancer patients seems to be low (around 2% of all cases) according to studies based on larger series of consecutive pancreatic cancer patients [18][19][45][46]. Most probably, this group needs a special treatment and could benefit from personalized treatment. Considering that 2% to 4% of all diagnosed cancers are mismatch repair-deficient, pancreatic cancer can fit into this range [47][48][49]. In general, microsatellite instability status represents a better prognostic factor for pancreatic cancer patients, potentially derived on a stronger anti-tumor response of the innate immune system [26].
Table 1. Major studies assessing MSI in pancreatic cancer.
| Author/Year | Study Population | Methodology | MSI in % |
|---|---|---|---|
| Luipinacci/2018 | 445 | IHC on resected samples from consecutive patients at multiple centers | 1.6 |
| Hu/2018 | 833 | NGS, PCR-based and IHC on resected samples from consecutive patients | 0.8 |
| Liu/2014 | 36 | IHC on resected and metastatic selected patients with acinar cell carcinoma | 13.8 |
| Abe/1996 | 44 | PCR based on resected samples | 15.9 |
| Yamamoto/2001 | 103 | PCR-based and IHC on resected samples from partially selected patients (3 Lynch Syndrome patients added to a series of 100 patients from multiple centers) | 15.5 |
| Abraham/2002 | 21 | PCR based on resected samples (17 patients) and core biopsies (4 patients) from selected patients with acinar cell carcinoma | 7.7 |
| Tomaszewska/2003 | 30 | IHC on resected samples from consecutive patients | 0.0 |
| Luttges/2003 | 23 | PCR-based and IHC on resected samples from selected patients (extensive invasive mucinous component) | 4.3 |
| Maple/2005 | 35 | PCR-based and IHC on selected patients (long-term survivors; ≥3 years) | 8.6 |
| Nakata/2002 | 46 | PCR based on resected samples from consecutive patients | 17.4 |
| Fujii/2009 | 21 | PCR based on resected samples | 0.0 |
| Laghi/2012 | 338 | PCR-based and IHC on samples from consecutive patients at multiple centers | 0.3 |
| Han/1993 | 9 | PCR based on resected samples | 67.0 |
| Seymour/1994 | 33 | PCR based on resected samples | 21.2 |
| Brentnall/1995 | 17 | PCR based on pancreatic juice | 75.0 |
| Venkatasubbarao/1998 | 14 | PCR based on resected samples | 57.0 |
| Ouyang/1997 | 51 | PCR based on resected samples | 14.0 |
| Ouyang/1998 | 60 | PCR based on resected samples | 15.0 |
| Goggins/1998 | 82 | PCR based on samples from consecutive patients | 3.7 |
| Ghimenti/1999 | 21 | PCR based on samples from consecutive patients | 67.0 |
| Wilentz/2000 | 18 | IHC and PCR based on samples from selected patients with medullary histology | 22.2 |
| Ueki/2000 | 36 | PCR based on resected samples | 11.1 |
| Nakata/2003 | 55 | IHC on resected samples | 9.2 |
| Ottenhof/2012 | 78 | IHC on resected samples from patients at multiple centers | 12.8 |
| Mitsuhaski/2015 | 283 | Methodology not specified | 0.0 |
| Riazy/2015 | 265 | IHC on resected samples from consecutive patients | 15.4 |
| Grant/2015 | 38 | NGS on blood samples from patients | 2.6 |
| Connor/2017 | 255 | NGS, PCR-based and IHC on resected samples (243 primary tumors and 12 metastases) | 1.7 |
| Lucchini/2020 | 8323 | Systematic review of 34 studies | 2.0 |
Legend: IHC: immunohistochemistry; NGS: next-generation sequencing; PCR: polymerase chain reaction.
For a long time, pancreatic cancers have been regarded as tumors that are able to evade a host immune system, but considering new studies this assessment has been modified. Following new observations, it is widely accepted now that immune cell infiltration is present in many cancers and it also might represent a prognostic tool in pancreatic cancer [50][51][52][53]. In this new view, pancreatic tumors are infiltrated by different subgroups of T-cells. High infiltration rate of CD4+ and CD8+ T-cells and low number of regulatory T-cells seems to be related to a better prognosis [54][55]. A recent study demonstrated that a high number of CD3+ and CD8+ T-cells are indicators of a more favorable prediction, moreover by combining them both into an immune cell score the prognostic value can be further improved [56]. It is known that in MSI pancreatic cancer the amount of CD8+ T-cells at the invasive front in addition to the expression level of PD-1 and PD-L1 is higher than in pancreatic cancer with intact mismatch repair system [18] (Figure 1). Nevertheless, the dense stromal tissue present in the tumor microenvironment might be the reason behind the wide variations in density of T-cells within the tumor area. Furthermore, compared to other tumors, pancreatic tumors are often characterized by a low level of activated cytotoxic CD8+ T-cells and an intense infiltration of immune-suppressive cells such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory CD4+ T-cells (Tregs) [53][57][58][59][60][61].
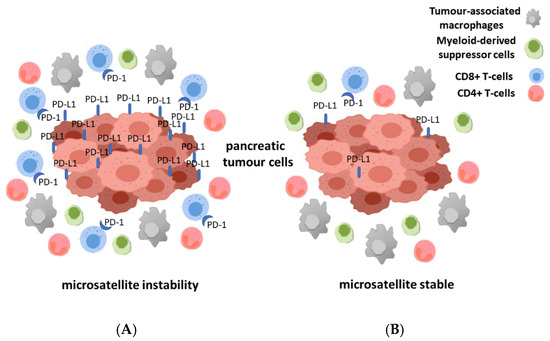
Figure 1. Illustration of the different immune milieu for microsatellite instable (MSI) (A) and microsatellite stabile (MSS) (B) pancreatic cancer. The different number of immune cells especially of CD8+ and CD4+ T-cells, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) in the tumor microenvironment of the two groups of pancreatic cancer is shown.
In contrast to colon cancer, there is currently no common consent regarding measuring immune response in pancreatic tumor [62]. A promising diagnostic advance in identifying MSI in pancreatic cancer patients has been done recently [63]. In this study, MSI was identified in liquid biopsies by testing circulating tumor DNA. This achievement overcomes the difficulties to obtain tumor tissue by means of traditional tissue biopsy from the pancreatic patients. Furthermore, in the same study, it was possible to monitor the effect of pembrolizumab targeted therapy by means of serial analysis of circulating tumor DNA. Recently, the advantage of using liquid biopsies for monitoring pancreatic cancer therapy has also been presented in a meta-analysis based on 19 studies including 1872 patients [64]. Therefore, analyzing circulating tumor DNA based on the non-invasive method of liquid biopsy could become a method to monitor the response to immune therapy in pancreatic cancer patients.
3. Chemotherapeutic Options in MSI Pancreatic Cancer Patients
Several studies with MSI colorectal cancer patients have shown that mismatch repair-deficient patients benefit differently from standard chemotherapy compared to microsatellite stable (MSS) ones, therefore 5-fluoruracil-based regimes for instance are not recommended for this colorectal cancer patient subgroup [65][66][67][68]. Additionally, pre-clinical and clinical studies provided evidence of elevated cytotoxic effects of some drugs in MSI tumors [69][70].
In general, mismatch repair deficiency alone is not a direct transforming factor for cells and only the subsequent accumulation of cancer specific further oncogenic mutations and other genomic alterations results in the develop of MSI tumors [71]. Moreover, on one hand, a deficient mismatch repair system might affect the malignancy through increased drug resistance (especially in case of methylating, alkylating, and platinum-containing agents). On the other hand, it results in a higher rate of potentially immunogenic neo-antigens and higher response rate to immune therapy [26][72][73]. Drug resistance mechanisms in mismatch repair deficient tumors can occur by an increased tolerance to DNA damage, reduced cell-cycle arrest ability and defective apoptotic signaling [73][74][75].
Therefore, in line with the above-described facts pancreatic cancer patients with MSI also react differently to chemotherapy, as documented by two different studies [42][76]. In one study, it was shown that pancreatic cancer patients react significantly differently towards an adjuvant chemotherapy with pyrimidine analogue. Patients with MSI have no survival advantage when treated with 5-fluoruracil or gemcitabine-based chemotherapy whereas patients with intact mismatch repair system have a 10-month-prolonged DFS [42]. In another study, metastatic pancreatic cancer patients with deficient mismatch repair system showed better outcome (median OS of 16.5 months) compared to patients with intact mismatch repair system (median OS of 11.1 months) while undergoing FOLFIRINOX treatment [76]. Both studies underline the benefit of a more personalized treatment for pancreatic cancer patients.
Nevertheless, in pancreatic tumors, as with in other gastrointestinal cancers (e.g., hepatocellular, gastric, renal cell, and esophageal cancer), the immune-suppressive tumor microenvironment and low immunogenicity of the tumor is a hurdle that must be overcome. Furthermore, pancreatic cancer has a low rate of somatic mutations and a minimal neo-epitope presentation [58][59][77]. Combination therapies to increase immune responsiveness seems to be one possibility to overcome this limitation. Results of early clinical studies using immune-checkpoint inhibition in pancreatic cancers have been disappointing so far [78].
Since 2017, the PD-1 (programmed cell death protein-1; CD279) inhibitor pembrolizumab is approved for treatment of mismatch repair-deficient cancers irrespective of the tumor site by the U.S. Food and Drug Administration [49][79]. In general, PD-L1 (programmed death-ligand 1; CD274) is expressed on the surface of cancer cells but only rarely expressed by normal tissues [80]. After binding of PD-1 to PD-L1 the proliferation of antigen-specific T-cells in lymph nodes is suppressed and apoptosis of Tregs is reduced. Therefore, antibodies targeting PD-1 and PD-L1 can restore anti-tumoral immunity by stimulating endogenous immune response [81].
Treatment based on pembrolizumab alone has been only successful in MSI-high (instability in at least two of the five microsatellites markers) pancreatic cancer patients but not in MSI-low (instability in only one of the five microsatellites markers) patients [82][83].
In some studies, anti-PD-1/PD-L1 molecules have been administered together with vaccine, conventional chemo- or radiotherapy for treatment of MSI-low pancreatic cancer with the aim to transform an immune-suppressive to an immune-active microenvironment [60][84][85][86][87].
Based on relevant in vivo experiments and a small clinical study with ten pancreatic cancer patients, it was concluded that chemotherapy with gemcitabine most probably has the capacity to enhance responses to immunotherapy [85][88]. Even if gemcitabine suppressed memory T-cells, it was able to increase naïve T-cell function [85]. In agreement with this observation, the combination therapy of gemcitabine with antibodies targeting PD-1 and PD-L1 induced a significant synergistic anti-tumor effect in mouse models of pancreatic tumor [86]. Phase I/II studies involving anti-PD-1 and PD-L1 in pancreatic cancer are summarized in Table 2.
Table 2. Phase I/II studies involving anti-PD-1 and PD-L1 in metastatic pancreatic cancer. [89][90][91][92][93][94][95][96][97]
| Author/Year | Study Phase | N° pts | N° MSI-H/PD-L1-H (%) | Anti-PD-1/PD-L1 Agent | Combination Agent | Outcomes |
|---|---|---|---|---|---|---|
| Weiss/2018 | I/II | 17 | 9 (53) MSI-H | pembro | gemcitabine/nab-paclitaxel | DCR 100%, mPFS 9.1 mo, mOS 15 mo |
| Wainberg/2020 | I | 50 | 12 (24) PD-L1 ≥ 1 | nivo | gemcitabine/nab-paclitaxel | mPFS 5.5 mo, mOS 9.9 mo |
| O’Reilly/2019 | II | 65 | 8 (12) PD-L1 ≥ 25 | durva | tremelimumab | DCR 9.4%, PR 3.1% |
| Tsujikawa/2020 | II | 93 | NA | nivo | Cy/GVAX/CRS-207 | mOS 5.9 mo |
| Borazanci/2018 | II | 25 | NA | nivo | paricalcitol, gemcitabine/nab-paclitaxel/cisplatin | PR 80%, DCR 100%, mPFS 8.2 mo, mOS NR |
| Wainberg/2017 | I | 31 | NA | nivo | cabiralizumab | ORR 13% in MSS |
| Calvo/2018 | I/II | 50 | NA | sparta | lacnotuzumab | DCR 46% |
| Wang-Gillam/2020 | I | 20 | NA | pembro | defactinib/ gemcitabine | mPFS 2.9 mo, mOS 7.6 mo |
| Hong/2019 | I/II | 49 | NA | durva | ibrutinib | mOS 4 mo |
| Overman/2018 | I | 20 | NA | durva | oleclumab | PR 10%, DCR 25% |
Legend: CRS-207: vaccine; Cy: cyclophosphamide; DCR: disease control rate; durva: durvalumab; GVAX: irradiated allogenic pancreatic tumor cells vaccine; mo: months; mOS: median overall survival; MSI-H: microsatellite high; mPFS: median progression-free survival; MSS: microsatellite stable; N°: number; NA: not available; nivo: nivolumab; NR: not reached; ORR: overall response rate; PD-L1-H: PD-L1 high; pembro: pembrolizumab; PR: partial response; sparta: spartalizumab.
17 patients were included in a phase I/II study and treated with gemcitabine, nab-paclitaxel, and pembrolizumab. The maximum tolerated dose of this treatment was pembrolizumab 2 mg/kg every 21 days, gemcitabine 1000 mg/m2 and nab-paclitaxel 125 mg/m2 on days 1 and 8 every 21 days. Among 11 evaluable patients, disease control rate (DCR) was 100%, median PFS was 9.1 months and OS 15 months [84]. In another phase I study the combination of nab-paclitaxel 125 mg/m2 and gemcitabine 1000 mg/m2 on days 1–8–15 were also tested with nivolumab at the dose of 3 mg/kg (days 1 and 15) on 50 patients. The combination was safe; however, activity beyond standard chemotherapy doublet was not registered, with a median PFS of 5.5 and median OS of 9.9 months [89]. In a study of second-line treatment for advanced pancreatic cancer after progression to 5-FU or gemcitabine-based regimens, 65 patients were randomized to monotherapy with durvalumab (1.5 g every 4 weeks) or the combination of durvalumab and tremelimumab (75 mg every 4 weeks). A partial response (PR) which persisted 9 months was observed in one patient (3.1%) in the combination arm and in 9.4% of patients’ disease control was reached. 6.1% of patients in the durvalumab alone arm demonstrated PR and disease control. Due to lack of efficacy signal demonstrated in the first part of the study, the trial was not further conducted to assess efficacy by overall response rate (ORR) [90].
In another attempt, a vaccine based on irradiated, allogenic pancreatic tumor cells expressing granulocyte-macrophage colony-stimulating factor (GVAX), was combined with PD-1L and PD-1 inhibitors. Combination of this vaccine with immune-checkpoint inhibitors was able to render pancreatic cancer accessible to immunotherapy [87]. In a phase II preliminary randomized study, all participants received two doses of low-dose cyclophosphamide to inhibit T-cells prior to GVAX vaccine activating a broad antigenic response (Cy/GVAX). Then, the patients were randomized between the prosecution of the Cy/GVAX protocol for six further cycles or switch to a different vaccine (CRS-207). CRS-207 is a vaccine of live-attenuated, mesothelin-expressing Listeria monocytogenes. Treatment with both vaccines was well-tolerated and combined treatment with CRS-207 and Cy/GVAX resulted in improved median OS of 6.1 months compared to 3.9 months for Cy/GVAX treatment alone [98]. The efficiency of these vaccines was analyzed further in a subsequent study (ECLIPSE study) based on 213 pancreatic cancer patients who received at least two prior treatment regimens. Patients were randomized and received either both vaccines (Cy/GVAX and CRS-207; arm A) together, or CRS207 alone (arm B), or single-agent chemotherapy according to physician’s choice (arm C). At the final analysis, there was no OS difference between the three treatment arms regarding median OS, with values of 3.7, 5.4 and 4.6 months, respectively. Thus, combining CRS-207 and Cy/GVAX vaccine resulted not in an improved survival over standard chemotherapy [99]. Another phase II study, the STELLAR study, included pancreatic cancer patients who received one prior treatment regimen. Ninety-three patients received first vaccination with Cy/GVAX followed by CRS-207 vaccine and were then randomized into two arms—either with or without nivolumab treatment. Every three weeks nivolumab was administered at a dose of 3 mg/kg for six total cycles together with Cy/GVAX followed by CRS-207. Median OS did not differ significantly between the two arms (5.9 months and 6.1 months, respectively) and the addition of nivolumab did not improve survival outcomes [91].
Vitamin D receptor agonist (paricalcitol) has demonstrated activity in sensitizing pancreatic cancer lesions to immune-checkpoint blockade by reducing the activity of MDSCs and Tregs [100]. In advanced chemotherapy-naïve pancreatic cancer patients, treatment with paricalcitol was combined with nivolumab, nab-paclitaxel, gemcitabine, and cisplatin. Preliminary results on ten treated patients reported partial regression in eight patients (80% of PR) and stable disease in two patients (100% of DCR). In this study, 8.2 months were indicated as median PFS and up to now no data for median OS are available [92].
MDSCs and TAMs account for the immunosuppressive microenvironment of pancreatic cancer [101]. TAMs and MDSCs are recruited to the tumor stroma by high levels of colony-stimulating factor 1 (CSF-1) secreted from pancreatic tumor cells [102]. Inhibition of the CSF-1 receptor resulted in reprogramming of TAM to M1 (more immunogenic cells), increased cytotoxic T-cell infiltration and reduced the amount of Tregs in the tumor microenvironment [103][104][105]. The CSF-1 receptor (CSF-1R) inhibitor cabiralizumab was administered together with nivolumab in a phase I study for pretreated advanced pancreatic cancer patients. Four out of 31 patients had an ORR of 13% and of special interest all belong to the MSS subgroup [104]. Another phase I/II trial tested the combination of lacnotuzumab (anti-CSF-1 monoclonal antibody) with spartalizumab (PD-1 inhibitor). From the 13 patients with advanced pancreatic cancer, six patients had disease control with this combination and three of six had a disease control superior to 300 days [104][105].
PD-1 inhibitors have been demonstrated to execute also synergistic effects with focal adhesion kinase (FAK) inhibitors. FAK is often overexpressed in pancreatic cancer and has well known oncogenic properties [104]. The FAK inhibitor defactinib was combined with pembrolizumab and gemcitabine in the frame of a phase I study. Among ten evaluable pancreatic cancer patients in maintenance treatment after chemotherapy with gemcitabine and nab-paclitaxel, 60% had stable disease and 10% progressed. 4.6 months was the median time on treatment. In the refractory cohort, included patients progressed to first-line chemotherapy, 50% had stable disease. Median progression-free disease was 2.9 months and median OS was 7.6 months. In the dose escalation cohort, one more patient progressed. Of interest, both progressing patients have been identified as MSS [95].
Ibrutinib, a bruton tyrosine kinase (BTK) inhibitor, plays a role in the immunomodulation of pancreatic cancer tumor microenvironment [106] and therefore the BTK-inhibitor ibrutinib was tested together with nab-paclitaxel and gemcitabine in advanced pancreatic cancer patients as first-line treatment (Resolve study) [107]. Furthermore, in a phase I/II study ibrutinib was also combined with PDL-1 inhibitor durvalumab but OS was poor with a median survival of 4 months only [96].
Another treatment strategy aimed at activating T-cells. One example is the use of AM0010, a pegylated IL-10. AM0010 was tested in second-line treatment of advanced pancreatic cancer in combination with FOLFOX (folinic acid, 5-fluorouracil (5-FU), oxaliplatin) chemotherapy. In this study, 25 participants were treated with AM0010 (5 ug/kg per day) in combination with FOLFOX (every 14 days). ORR was 15.8% with a DCR of 78.9%. Median PFS was 3.5 months, median OS was 10.2 months and 1-year survival 43% [108]. The subsequent SEQUOIA trial (phase III) tested the combination of pegylated IL-10 (pegilodecakin) together with FOLFOX in gemcitabine refractory pancreatic cancer patients. The control arm received only chemotherapy with FOLFOX. Surprisingly, the addition of pegilodecakin to FOLFOX did not improve efficacy (PFS, OR, ORR) [109].
A different target is CD73, a cell surface enzyme often up-regulated in pancreatic cancer. CD73 exerts an immunosuppressive effect by generating extracellular adenosine [110]. Oleclumab, a human monoclonal antibody that binds to CD73, was tested in combination with durvalumab. As expected, production of immunosuppressive adenosine was reduced and the amount of CD8+ T-cells in the tumor microenvironment increased after oleclumab treatment. Partial regression was observed in 10% of cases, while DCR was present in 25% of patients [97]. Another enzyme with immunosuppressive effect is indoleamine-2,3-dioxygenase-1 (IDO1) [111]. Increased IDO-1 expression on tumor cells results in NK-cell and T-cell suppression, Tregs activation and promotion of immune tolerance [112]. In a phase II study, 135 patients were evaluated for first-line treatment with nab-paclitaxel, gemcitabine, and the IDO1 inhibitor indoximod. In this study, the reported median OS was 10.2 months and ORR was 46.2%, with 45.2% of partial repression. The intratumoral CD8+ T-cell density was higher in responders of this treatment than in non-responders [113].
References
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W.; American College of G. ACG clinical guide-line: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262, doi:10.1038/ajg.2014.435.
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: Molecular bases, clinical perspectives, and new treatment approaches. Cell Mol. Life Sci. 2018, 75, 4151–4162, doi:10.1007/s00018-018-2906-9.
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103, doi:10.1073/pnas.1015541107.
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987, doi:10.1073/pnas.1002472107.
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009, 302, 1790–1795, doi:10.1001/jama.2009.1529.
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, mo-lecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18, doi:10.1111/j.1399-0004.2009.01230.x.
- Humphris, J.L.; Patch, A.M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation In Pancreatic Cancer. Gastroenterology 2017, 152, 68–74, doi:10.1053/j.gastro.2016.09.060.
- FCDAS; Wernhoff, P.; Dominguez-Barrera, C.; Dominguez-Valentin, M. Update on Hereditary Colorectal Cancer. Anticancer Res. 2016, 36, 4399–4405, doi:10.21873/anticanres.10983.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413, doi:10.1126/science.aan6733.
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337, doi:10.1038/nature11252.
- Bujanda, L.; Herreros-Villanueva, M. Pancreatic Cancer in Lynch Syndrome Patients. J. Cancer 2017, 8, 3667–3674, doi:10.7150/jca.20750.
- Banville, N.; Geraghty, R.; Fox, E.; Leahy, D.T.; Green, A.; Keegan, D.; Geoghegan, J.; O’Donoghue, D.; Hyland, J.; Sheahan, K. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum. Pathol. 2006, 37, 1498–1502, doi:10.1016/j.humpath.2006.06.024.
- Grover, S.; Syngal, S. Hereditary pancreatic cancer. Gastroenterology 2010, 139, 1076–1080, doi:10.1053/j.gastro.2010.08.012.
- Geary, J.; Sasieni, P.; Houlston, R.; Izatt, L.; Eeles, R.; Payne, S.J.; Fisher, S.; Hodgson, S.V. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam. Cancer 2008, 7, 163–172, doi:10.1007/s10689-007-9164-6.
- Liu, W.; Shia, J.; Gonen, M.; Lowery, M.A.; O’Reilly, E.M.; Klimstra, D.S. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: Frequency and clinical significance. Pancreas 2014, 43, 1264–1270, doi:10.1097/MPA.0000000000000190.
- Karamurzin, Y.; Zeng, Z.; Stadler, Z.K.; Zhang, L.; Ouansafi, I.; Al-Ahmadie, H.A.; Sempoux, C.; Saltz, L.B.; Soslow, R.A.; O’Reilly, E.M.; et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: A report of new cases and review of the literature. Hum. Pathol. 2012, 43, 1677–1687, doi:10.1016/j.humpath.2011.12.012.
- Lee, S.H.; Kim, W.Y.; Hwang, D.Y.; Han, H.S. Intraductal papillary mucinous neoplasm of the ileal heterotopic pancreas in a patient with hereditary non-polyposis colorectal cancer: A case report. World J. Gastroenterol. 2015, 21, 7916–7920, doi:10.3748/wjg.v21.i25.7916.
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Marechal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gas-troenterology 2018, 154, 1061–1065, doi:10.1053/j.gastro.2017.11.009.
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336, doi:10.1158/1078-0432.CCR-17-3099.
- Abe, T.; Ouyang, H.; Migita, T.; Kato, Y.; Kimura, M.; Shiiba, K.; Sunamura, M.; Matsuno, S.; Horii, A. The somatic mutation frequency of the transforming growth factor β receptor type II gene varies widely among different cancers with microsatellite instability. Eur. J. Surg. Oncol. 1996, 22, 474–477, doi:10.1016/s0748-7983(96)92824-3.
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of hu-man pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144.
- Abraham, S.C.; Wu, T.T.; Hruban, R.H.; Lee, J.H.; Yeo, C.J.; Conlon, K.; Brennan, M.; Cameron, J.L.; Klimstra, D.S. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: Frequent allelic loss on chromosome 11p and altera-tions in the APC/β-catenin pathway. Am. J. Pathol. 2002, 160, 953–962, doi:10.1016/s0002-9440(10)64917-6.
- Tomaszewska, R.; Okon, K.; Stachura, J. Expression of the DNA mismatch repair proteins (hMLH1 and hMSH2) in infiltrat-ing pancreatic cancer and its relation to some phenotypic features. Pol. J. Pathol. 2003, 54, 31–37.
- Luttges, J.; Beyser, K.; Pust, S.; Paulus, A.; Ruschoff, J.; Kloppel, G. Pancreatic mucinous noncystic (colloid) carcinomas and intraductal papillary mucinous carcinomas are usually microsatellite stable. Mod. Pathol. 2003, 16, 537–542, doi:10.1097/01.MP.0000072748.65178.2F.
- Maple, J.T.; Smyrk, T.C.; Boardman, L.A.; Johnson, R.A.; Thibodeau, S.N.; Chari, S.T. Defective DNA mismatch repair in long-term (> or =3 years) survivors with pancreatic cancer. Pancreatology 2005, 5, 220–227, doi:10.1159/000085275.
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Nishioka, N.; Tanaka, H.; Ohira, M.; Ishikawa, T.; Nishino, H.; Hirakawa, K. Prognos-tic value of microsatellite instability in resectable pancreatic cancer. Clin. Cancer Res. 2002, 8, 2536–2540.
- Fujii, K.; Miyashita, K.; Yamada, Y.; Eguchi, T.; Taguchi, K.; Oda, Y.; Oda, S.; Yoshida, M.A.; Tanaka, M.; Tsuneyoshi, M. Simulation-based analyses reveal stable microsatellite sequences in human pancreatic cancer. Cancer Genet. Cytogenet. 2009, 189, 5–14, doi:10.1016/j.cancergencyto.2008.09.008.
- Laghi, L.; Beghelli, S.; Spinelli, A.; Bianchi, P.; Basso, G.; Di Caro, G.; Brecht, A.; Celesti, G.; Turri, G.; Bersani, S.; et al. Irrele-vance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e46002, doi:10.1371/journal.pone.0046002.
- Han, H.J.; Yanagisawa, A.; Kato, Y.; Park, J.G.; Nakamura, Y. Genetic instability in pancreatic cancer and poorly differenti-ated type of gastric cancer. Cancer Res. 1993, 53, 5087–5089.
- Seymour, A.B.; Hruban, R.H.; Redston, M.; Caldas, C.; Powell, S.M.; Kinzler, K.W.; Yeo, C.J.; Kern, S.E. Allelotype of pan-creatic adenocarcinoma. Cancer Res. 1994, 54, 2761–2764.
- Brentnall, T.A.; Chen, R.; Lee, J.G.; Kimmey, M.B.; Bronner, M.P.; Haggitt, R.C.; Kowdley, K.V.; Hecker, L.M.; Byrd, D.R. Microsatellite instability and K-ras mutations associated with pancreatic adenocarcinoma and pancreatitis. Cancer Res. 1995, 55, 4264–4267.
- Venkatasubbarao, K.; Ahmed, M.M.; Swiderski, C.; Harp, C.; Lee, E.Y.; McGrath, P.; Mohiuddin, M.; Strodel, W.; Freeman, J.W. Novel mutations in the polyadenine tract of the transforming growth factor β type II receptor gene are found in a sub-population of human pancreatic adenocarcinomas. Genes Chromosomes Cancer. 1998, 22, 138–144, doi:10.1002/(sici)1098-2264(199806).
- Ouyang, H.; Shiwaku, H.O.; Hagiwara, H.; Miura, K.; Abe, T.; Kato, Y.; Ohtani, H.; Shiiba, K.; Souza, R.F.; Meltzer, S.J.; et al. The insulin-like growth factor II receptor gene is mutated in genetically unstable cancers of the endometrium, stomach, and colorectum. Cancer Res. 1997, 57, 1851–1854.
- Ouyang, H.; Furukawa, T.; Abe, T.; Kato, Y.; Horii, A. The BAX gene, the promoter of apoptosis, is mutated in genetically unstable cancers of the colorectum, stomach, and endometrium. Clin. Cancer Res. 1998, 4, 1071–1074.
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic his-topathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507.
- Ghimenti, C.; Tannergard, P.; Wahlberg, S.; Liu, T.; Giulianotti, P.G.; Mosca, F.; Fornaciari, G.; Bevilacqua, G.; Lindblom, A.; Caligo, M.A. Microsatellite instability and mismatch repair gene inactivation in sporadic pancreatic and colon tumours. Br. J. Cancer 1999, 80, 11–16, doi:10.1038/sj.bjc.6690314.
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651, doi:10.1016/S0002-9440(10)65035-3.
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancre-atic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839.
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Ohira, M.; Ishikawa, T.; Nishino, H.; Seki, S.; Hirakawa, K. Negative hMSH2 protein expression in pancreatic carcinoma may predict a better prognosis of patients. Oncol. Rep. 2003, 10, 997–1000.
- Ottenhof, N.A.; Morsink, F.H.; Ten Kate, F.; van Noorden, C.J.; Offerhaus, G.J. Multivariate analysis of immunohistochemi-cal evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent Smad4 expression only. Cell Oncol. 2012, 35, 119–126, doi:10.1007/s13402-012-0072-x.
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220, doi:10.18632/oncotarget.3109.
- Riazy, M.; Kalloger, S.E.; Sheffield, B.S.; Peixoto, R.D.; Li-Chang, H.H.; Scudamore, C.H.; Renouf, D.J.; Schaeffer, D.F. Mis-match repair status may predict response to adjuvant chemotherapy in resectable pancreatic ductal adenocarcinoma. Mod. Pathol. 2015, 28, 1383–1389, doi:10.1038/modpathol.2015.89.
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenter-ology 2015, 148, 556–564, doi:10.1053/j.gastro.2014.11.042.
- Connor AA.; Denroche RE.; Jang GH.; Timms, L.; Kalimuthu SN.; Selander, I.; McPherson, T.; Wilson GW.; Chan-Seng-Yue MA.; Borozan, I.; Ferretti, V.; Grant RC.; Lungu IM.; Costello, E.; Greenhalf, W.; Palmer, D.; Ghaneh, P.; Neoptolemos JP.; Buchler, M.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783, doi:10.1001/jamaoncol.2016.3916.
- Cancer Genome Atlas Research Network. Electronic address aadhe; Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203, doi:10.1016/j.ccell.2017.07.007.
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: His-tology, molecular pathology and clinical implications. Gut 2020, doi:10.1136/gutjnl-2020-320726.
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180, doi:10.1038/ncomms15180.
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Land-scape of Microsatellite Instability Across 39 Cancer Types. JCO Precis Oncol. 2017, 2017, doi:10.1200/PO.17.00073.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520, doi:10.1056/NEJMoa1500596.
- Cai, S.W.; Yang, S.Z.; Gao, J.; Pan, K.; Chen, J.Y.; Wang, Y.L.; Wei, L.X.; Dong, J.H. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery 2011, 149, 576–584, doi:10.1016/j.surg.2010.10.009.
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551, doi:10.1371/journal.pone.0091551.
- Jamieson, N.B.; Mohamed, M.; Oien, K.A.; Foulis, A.K.; Dickson, E.J.; Imrie, C.W.; Carter, C.R.; McKay, C.J.; McMillan, D.C. The relationship between tumor inflammatory cell infiltrate and outcome in patients with pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2012, 19, 3581–3590, doi:10.1245/s10434-012-2370-y.
- Yoshikawa, K.; Mitsunaga, S.; Kinoshita, T.; Konishi, M.; Takahashi, S.; Gotohda, N.; Kato, Y.; Aizawa, M.; Ochiai, A. Im-pact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012, 103, 2012–2020, doi:10.1111/j.1349-7006.2012.02411.x.
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indi-cator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923, doi:10.1038/bjc.2013.32.
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095, doi:10.1038/ncomms15095.
- Tahkola, K.; Mecklin, J.P.; Wirta, E.V.; Ahtiainen, M.; Helminen, O.; Bohm, J.; Kellokumpu, I. High immune cell score pre-dicts improved survival in pancreatic cancer. Virchows Arch. 2018, 472, 653–665, doi:10.1007/s00428-018-2297-1.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276, doi:10.1158/1078-0432.CCR-11-3114.
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017, 17, 68, doi:10.1186/s12935-017-0437-3.
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancre-atic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240, doi:10.1053/j.gastro.2012.12.042.
- Johnson, B.A., 3rd; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogen-icity. Clin. Cancer Res. 2017, 23, 1656–1669, doi:10.1158/1078-0432.CCR-16-2318.
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immu-nosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136, doi:10.1136/gutjnl-2016-312078.
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell HK.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; Nag-tegaal ID.; Palmqvist, R.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209, doi:10.1002/path.4287.
- Kamatham, S.; Shahjehan, F.; Kasi PM. Circulating Tumor DNA-Based Detection of Microsatellite Instability and Response to Immunotherapy in Pancreatic Cancer. Front. Pharmacol. 2020, 11, 23, doi:10.3389/fphar.2020.00023.
- Zhu, Y.; Zhang, H.; Chen, N.; Hao, J.; Jin, H.; Ma, X. Diagnostic value of various liquid biopsy methods for pancreatic can-cer: A systematic review and meta-analysis. Medicine 2020, 99, e18581, doi:10.1097/MD.0000000000018581.
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004, 126, 394–401, doi:10.1053/j.gastro.2003.12.023.
- Benatti, P.; Gafa, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite instability and colorectal cancer prognosis. Clin. Cancer Res. 2005, 11, 8332–8340, doi:10.1158/1078-0432.CCR-05-1030.
- De Vos tot Nederveen Cappel, W.H.; Meulenbeld, H.J.; Kleibeuker, J.H.; Nagengast, F.M.; Menko, F.H.; Griffioen, G.; Cats, A.; Morreau, H.; Gelderblom, H.; Vasen, H.F. Survival after adjuvant 5-FU treatment for stage III colon cancer in hereditary nonpolyposis colorectal cancer. Int. J. Cancer 2004, 109, 468–471, doi:10.1002/ijc.11712.
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Pinol, V.; Xicola, R.M.; Bujanda, L.; Rene, J.M.; et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut 2006, 55, 848–855, doi:10.1136/gut.2005.073015.
- Jacob, S.; Aguado, M.; Fallik, D.; Praz, F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomer-ase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001, 61, 6555–6562.
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.C.; Ducreux, M.; Praz, F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003, 63, 5738–5744.
- Duval, A.; Hamelin, R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: Toward a new concept of target genes for instability. Cancer Res. 2002, 62, 2447–2454.
- Sinicrope, F.A.; Sargent, D.J. Molecular pathways: Microsatellite instability in colorectal cancer: Prognostic, predictive, and therapeutic implications. Clin. Cancer Res. 2012, 18, 1506–1512, doi:10.1158/1078-0432.CCR-11-1469.
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6.
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346, doi:10.1038/nrm1907.
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2’,2’-difluorodeoxycytidine (gemcitabine). Drug Re-sist. Updat. 2002, 5, 19–33, doi:10.1016/s1368-7646(02)00002-x.
- Cloyd, J.M.; Katz, M.H.G.; Wang, H.; Cuddy, A.; You, Y.N. Clinical and Genetic Implications of DNA Mismatch Repair De-ficiency in Patients With Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2017, 152, 1086–1088, doi:10.1001/jamasurg.2017.2631.
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792, doi:10.1038/cddis.2015.162.
- Rosenberg, A.; Mahalingam, D. Immunotherapy in pancreatic adenocarcinoma-overcoming barriers to response. J. Gastroin-test Oncol. 2018, 9, 143–159, doi:10.21037/jgo.2018.01.13.
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412, doi:10.1056/NEJMp1709968.
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al Tu-mor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800, doi:10.1038/nm730.
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673, doi:10.1002/cam4.106.
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465, doi:10.1056/NEJMoa1200694.
- Patnaik, A.; Kang SP.; Rasco, D.; Papadopoulos KP.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Es-pino, G.; Gergich, K.; Delgado, L.; et al. Phase I Study of Pembrolizumab (MK-3475, Anti-PD-1 Monoclonal Antibody) in Pa-tients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293, doi:10.1158/1078-0432.CCR-14-2607.
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gem-citabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. N. Drugs 2018, 36, 96–102, doi:10.1007/s10637-017-0525-1.
- Plate, J.M.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of gemcitabine on immune cells in subjects with adenocarci-noma of the pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925, doi:10.1007/s00262-004-0638-1.
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in hu-man pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157, doi:10.1158/1078-0432.CCR-06-2746.
- Lutz ER.; Wu AA.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; Laheru, D.; Wolfgang CL.; Wang, J.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of im-mune regulation. Cancer Immunol. Res. 2014, 2, 616–631, doi:10.1158/2326-6066.CIR-14-0027.
- Nowak, A.K.; Robinson, B.W.; Lake, R.A. Synergy between chemotherapy and immunotherapy in the treatment of estab-lished murine solid tumors. Cancer Res. 2003, 63, 4490–4496.
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822, doi:10.1158/1078-0432.CCR-20-0099.
- O’Reilly EM.; Oh DY.; Dhani, N.; Renouf DJ.; Lee MA.; Sun, W.; Fisher, G.; Hezel, A.; Chang SC.; Vlahovic, G.; Takahashi, O.; Yang, Y.; Fitts, D.; Philip PA. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Duc-tal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, doi:10.1001/jamaoncol.2019.1588.
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Lahe-ru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588, doi:10.1158/1078-0432.CCR-19-3978.
- Borazanci, E.; Jameson, G.S.; Borad, M.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Von Hoff, D.D. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36, doi:10.1200/JCO.2018.36.4_suppl.358].
- Wainberg, Z.A.; Piha-Paul, S.A.; Luke, J.J. First-inhuman phase 1 dose escalation and expansion of a novel combination, an-ti–CSF-1 receptor (cabiralizumab) plus anti–PD-1 (nivolumab), in patients with advanced solid tumors. In Proceedings of the 32nd SITC Annual Meeting, National Harbor, MD, USA, 8–12 November 2017,
- Calvo, A.; Joensuu, H.; Sebastian, M. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J. Clin. Oncol. 2018, 36, S3014.
- Wang-Gillam, A.; McWilliams, R.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.-H.; Pedersen, K.S.; Trikalinos, N.; Aranha, O.; Park, H.S.; et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer: Experiences of pancreatic ductal adenocarcinoma (PDAC) patients. Cancer Res. 2020, 80, CT118, doi:10.1158/1538-7445.AM2020-CT118.
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Pa-tients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111, doi:10.1159/000500571.
- Overman, M.J.; Lorusso, P.; Strickler, J.H.; Patel, S.P.; Clarke, S.J.; Noonan, A.M.; Prasanna, T.; Amin, M.A.; Nemunaitis, J.J.; Desai, J.; et al. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with dur-valumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J. Clin. Oncol. 2018, 36, doi:10.1200/JCO.2018.36.15_suppl.4123.
- Whiting, C.; Lutz, E.; Nair, N.; Chang, S.C.; Lemmens, V.E.; Chen, S.-J.; Solt, S.; Ferber, S.; Maecker, H.; Murphy, A.; Brock-stedt, D.G.; Jaffee, E.M.; Le, D.T. Phase II, randomized study of GVAX pancreas and CRS-207 immunotherapy in patients with metastatic pancreatic cancer: Clinical update on long term survival and biomarker correlates to overall survival. J. Clin. Oncol. 2015, 33 (Suppl. 261), doi:10.1200/jco.2015.33.3_suppl.261.
- Le DT.; Picozzi VJ.; Ko AH.; Wainberg ZA.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse MA.; Zeh HJ, 3rd; Weekes, C.; Reid, T.; Borazanci, E.; Crocenzi, T.; LoConte NK.; Musher, B.; Laheru, D.; Murphy, A.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502, doi:10.1158/1078-0432.CCR-18-2992.
- Perez, K.; Cleary, J.M.; Karasic, T.B.; Raghavan, S.; Rahma, O.E.; Nowak, J.; Borazanci, E.; Downes, M.; Drebin, J.A.; Tuveson, D.A.; et al. Vitamin D receptor agonist paricalcitol plus gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2020, 38, doi:10.1200/JCO.2020.38.4_suppl.TPS784.
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in on-cology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416, doi:10.1038/nrclinonc.2016.217.
- Steins, A.; van Mackelenbergh, M.G.; van der Zalm, A.P.; Klaassen, R.; Serrels, B.; Goris, S.G.; Kocher, H.M.; Waasdorp, C.; de Jong, J.H.; Tekin, C.; et al. High-grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF-1. EMBO Rep. 2020, 21, e48780, doi:10.15252/embr.201948780.
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069, doi:10.1158/0008-5472.CAN-13-3723.
- Das, S.; Berlin, J.; Cardin, D. Harnessing the Immune System in Pancreatic Cancer. Curr. Treat. Options Oncol. 2018, 19, 48, doi:10.1007/s11864-018-0566-5.
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860, doi:10.1038/nm.4123.
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer. 2018, 17, 57, doi:10.1186/s12943-018-0779-z.
- Tempero, M.A.; Oh, D.Y.; Macarulla, T.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.; Starling, N.; Bachet, J.; Chang, H.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine as first-line treatment for patients with meta-static pancreatic adenocarcinoma: Results from the phase 3 RESOLVE study. Ann. Oncol. 2019, doi:10.1093/annonc/mdz154.001.
- Hecht, J.R.; Naing, A.; Falchook, G.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Lee Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Javle, M.; et al. Phase 1b study with PEGylated human IL-10 (AM0010) with 5-FU and oxaliplatin (FOLFOX) in metastatic pancreatic adenocarcinoma (PDAC). J. Clin. Oncol. 2017, 35, doi:10.1200/JCO.2017.35.4_suppl.399.
- Hecht JR.; Lonardi, S.; Bendell JC.; Sim, H.-W.; Macarulla, T.; Lopez CD.; Van Cutsem, E.; Munoz Martin AJ.; Park JO.; Grell, R.; Lin, Y.; Rao DS.; Ryoo B-Y. Randomized Phase III Study of FOLFOX Alone and with Pegilodecakin as Second-line Thera-py in Patients with Metastatic Pancreatic Cancer (SEQUOIA). J. Clin. Oncol. 2020, 38, doi:10.1200/JCO.2020.38.4_suppl.637.
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255, doi:10.1158/0008-5472.CAN-09-3109].
- Zhang, T.; Tan, X.L.; Xu, Y.; Wang, Z.Z.; Xiao, C.H.; Liu, R. Expression and Prognostic Value of Indoleamine 2,3-dioxygenase in Pancreatic Cancer. Chin. Med. J. 2017, 130, 710–716, doi:10.4103/0366-6999.201613.
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bed-side. J. Hematol. Oncol. 2018, 11, 100, doi:10.1186/s13045-018-0644-y.
- Bahary, N.; Garrido-Laguna, I.; Wang-Gillam, A.; Nyak-Kapoor, A.; Kennedy, E.; Vahanian, N.N.; Link, C.J. Results of the phase Ib portion of a phase I/II trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcita-bine/nab-paclitaxel for the treatment of metastatic pancreatic cancer. J. Clin. Oncol. 2016, 34, doi:10.1200/jco.2016.34.4_suppl.452.

