 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eva María Jiménez-Mateos | + 2544 word(s) | 2544 | 2020-12-18 09:25:08 | | | |
| 2 | Lily Guo | Meta information modification | 2544 | 2021-01-20 12:33:45 | | |
Video Upload Options
Neuroinflammation caused by hypoxia or hypoxic ischemia during the perinatal period contributes to increased risk for neurological deficits and long-term disabilities in children. Inflammation induced by injury results in activation of the resident and peripheral immune cells and production of cytokines.
1. Inflammation in Neonatal Encephalopathy
Neuroinflammation caused by hypoxia or hypoxic ischemia during the perinatal period contributes to increased risk for neurological deficits and long-term disabilities in children[1]. Inflammation induced by injury results in activation of the resident and peripheral immune cells and production of cytokines. In recent years, inflammation has been implicated in neonatal brain damage following perinatal stress. Induction and activation of microglia and astrocytes are hallmarks of neuroinflammation, which occurs in response to hypoxia and hypoxic ischemia in neonates[2][3]. Studies have shown that exposure of the neonatal brain to hypoxia, thereby causing an inflammatory response, is associated with long-lasting changes to neuronal morphology within the hippocampus and other vulnerable structures of the brain[4]. Circulating cytokines and neuroimmune cells such as microglia are the targets of several studies that are investigating the potential use of these components as biomarkers for neonatal brain injury. Additionally, targeting inflammation improves acute and long-lasting effects after hypoxia and hypoxic ischemia in mice [2][3].
Studies into the inflammatory response post-hypoxia have revealed that HIE is a sexually dimorphic disease, with male infants being far more vulnerable to ischaemic insults. Male infants are also significantly more at risk of suffering from long-term cognitive deficits when compared to female infants with comparable brain damage[5]. In fact, the microglial anti-inflammatory response was more robust in females than in males[5] [72]. More infiltration of peripheral lymphocytes, along with upregulated TNFα and IL10, were observed in males when compared with females. It was also noted that neurogenesis was more highly induced in female HIE brains versus male HIE brains. The conclusions drawn from this study were that the pro- and anti-inflammatory responses are indeed dichotomous with respect to sex, which is integral to the sex-specific chronic HIE outcomes, and that increased induction of neurogenesis in females also contributes to this sex-specific difference[5].
It has been long established that infants born prematurely are more vulnerable to the effects of asphyxia and developing adverse sequelae. Acute asphyxia at birth followed by HIE is more frequently seen in infants born prematurely than infants born at term, and is highly associated with the onset of adverse neurological outcomes[6]. Several studies have found that while HIE still only occurs in a minority of preterm births, it is a significant contributor to severe disability[7][8][9]. Mild HIE in preterm infants can result in white matter injury, even in the absence of abnormalities in neurological exams at discharge. The pattern of injury onset can be more prolonged in preterm infants; in some patients who develop cerebral palsy, there are no white matter lesions; however, long-term studies showed that it may be delayed onset demyelination [10]. As a result of this white matter injury, even in mild cases, the resultant adverse outcomes can be attributed to impaired global and regional connectivity between cortical and subcortical grey matter structures [11]. Evidence has also shown a correlation between preterm births and stunted cortical plasticity in adolescents [12]. Extensive evidence from preclinical studies has strongly implicated CNS and peripheral immune responses in the pathogenesis of HIE and preterm brain injury [1]. Various clinical and human post-mortem studies have shown that chronically upregulated systemic and CNS cytokines and gliosis show strong associations with adverse neurological outcomes [13][14]. Evidence has shown that systemic upregulation of TNFα and IL1β in premature infants is associated with impaired neural functions in the first 72 h of life, followed by cognitive impairment at 2 to 3 years of age [14].
It is clear from both human and animal studies that inflammation and the immune response are key to many aspects of the pathogenesis and pathophysiology of HIE and neonatal brain damage. However, further studies will be necessary to elucidate the underlying mechanisms associated with neonatal brain damage.
2. Pathogen-Associated Molecular Patterns (PAMPS) and Damage-Associated Molecular Patterns (DAMPS)
Pathogen-associated molecular patterns, or PAMPs, are normally conserved microbial products such as lipopolysaccharides which activate pattern recognition receptors (PRRs. such as Toll-like receptors (TLRs)) after a bacterial or viral infection. PRR signalling pathways have been shown to initiate cascades that lead to immune cells recruitment to the site of an infection [15]. In contrast to this, danger-associated molecular patterns, or DAMPs, are molecular patterns associated with sterile inflammation, or inflammation instigated without introduction of a pathogenic microbe, as seen after neuronal necrosis. These molecular patterns are released in response to tissue damage, with the same innate pattern recognition systems used in the detection of microbes initiating this sterile inflammatory response [15]. This inflammatory response, followed by tissue repair, is dependent on microglia migration to and from the site of injury [16]. Studies show that DAMPs and PAMPs induce distinctly different inflammatory responses in the neonatal brain (Figure 1). Lalancette-Hebert and colleagues [15] studied the difference between these responses with respect to toll-like receptor 2 (TLR2) expression. It was found that a neonatal mouse model of infection induced TLR2 expression and secretion of inflammatory mediators. Contrasting results were seen in the two neonatal mouse models of sterile inflammation (IL-1β injection and MCAO), which showed decreased induction of TLR2 and reduced production of inflammatory cytokines [17]. This study highlights the existence of scenario-specific innate immune responses, depending on the presence of either infections or sterile inflammation, and the necessity of looking for specific therapeutic strategies depending on the original insult. In the following sections we will examine the activation of the most studied family of receptors in hypoxia and hypoxia–ischemia murine animal models.
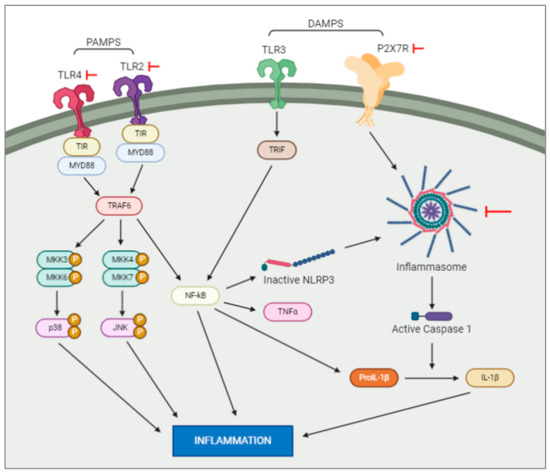
Figure 1. Intracellular pathways activated by DAMPS and PAMPS. DAMPS and PAMPS will bind to the membrane receptors, e.g., TLR and P2X7. This activation will trigger a series of intracellular events resulting in an increase of inflammation. Note: the red arrows represent experiments which have blocked the actions of the elements they are pointed at.
3. Toll-Like Receptors in Neonatal Encephalopathy
Upregulation of pattern-recognition receptors, such as TLRs, has been shown after perinatal brain injury. The Toll like receptor (TLR) family consists of nine subtypes (TLR1–9). They recognize a variety of pathogen-associated molecular patterns (PAMPS), including LPS, bacterial DNA and double stranded RNA. The role of TLR in perinatal brain injury has been extensively studied. TLR1, 2 and 7 are up-regulated 24 h after hypoxic ischemia in pups. TLR5 is downregulated and TLR3, 4, 6, 8 and 9 do not change expression. Interestingly, when KO mice for TLR 1 and 2 were subjected to hypoxic ischemia, TLR2 KO improved the infarct volume after hypoxic ischemia, but TLR1 KO did not have an effect. This data showed that TLR2 plays a role in initiating inflammation after perinatal brain injury [18][19][20]. Supporting this data, the use of Candesartan Cilexetil, a drug which reduces levels of TLR2, has been shown to improve neuronal damage after hypoxia in mice, and also improve the long-lasting neurological outcomes [2]. Importantly, the response of TLR2 to sterile (e.g., HI) or non-sterile inflammation (e.g., LPS to mimic bacterial infection) differs on the pre-clinical model [17]. Suggesting that, deeper knowledge of the pathways underlying TLR2 is important for developing new pharmacological treatments.
TLR4, another receptor implicated in DAMP and PAMP functions, has also been studied, due to observations that its inhibition has neuroprotective effects in neonatal brain damage[20]. TLR4 inhibition shortly after injury reduced activation of hippocampal glial cells improved hippocampal neuronal loss later in life and resulted in less severe long-term neurological outcomes [20]. These studies, by targeting receptors which mediate the effects of DAMPs and PAMPs, show the important roles played by these molecules in the mediation of the neuroinflammatory response following hypoxic ischaemic brain injury. Further studies are needed to fully understand these interactions in the neonatal brain post-injury.
4. Purinergic Signalling Activation after Neonatal Encephalopathy
Extracellular adenosine triphosphate (ATP) is a typical DAMP which acts as a glio- and neurotransmitter in the CNS to modulate functions such as brain excitability and neuroinflammation [21]. It is considered to be a co-transmitter in most neurons of the central and peripheral nervous system, and is released from astrocytes and neurons to act as either a co-transmitter or a sole transmitter[22]. The P2X class of ionotropic receptors, made up of seven distinct receptors, mediates the rapid effects of extracellular ATP by gating sodium and calcium entry into cells [23]. The P2X7 receptor (P2X7R) modulates cytokine production, glial activity and neurotransmitter release following brain injury [23]. P2X7R activation is seen in instances of pathologically high extracellular ATP levels, the likes of which are seen during seizures and brain injury. Downstream signalling of the P2X7R results in microglia activation and the release of interleukin 1β (IL-1β), which is a pro-convulsive inflammatory cytokine [24][25][26]. Evidence has shown that P2X7R is expressed by neurons and acts as a modulator of neurotransmitter release [27][28]. Similarly, each member of the P2Y class of eight purinergic metabotropic receptors is stimulated by ATP, and they are generally associated with slower presynaptic functions, and mediation of trophic signalling in cell differentiation, proliferation and death during development [22]. During epileptic seizures, large quantities of nucleotides enter the extracellular space from neurons and glia due to metabolic limitations [29]. These activate the P2X and P2Y receptors, including P2X7 and P2Y1, which are expressed on both embryonic and adult neural progenitor cells (NPCs). These two receptors regulate NPC functions, causing necrosis and apoptosis, and proliferation, differentiation and migration [30][31]. In a study by Rozmer and colleagues [32], patch-clamp recordings were carried out on hippocampal brain slices from neonate and adult transgenic nestin reporter mice which underwent pilocarpine-induced status epilepticus. This study detected the presence of P2X7R in NPCs in the subgranular zone of the dentate gyrus. Upon activation of these receptors, inward current was recorded near the resting membrane potential of the NPCs. P2Y1 receptor activation, on the other hand, initiated outward current close to the reverse potential of the P2X7R current [32]. It was also noted that the sensitivity of these two receptors was invariably increased. In this model, status epilepticus was preceded by a latency of 5 days after treatment with pilocarpine, and recurrent epileptic fits occurred during this period. Blockade of central P2X7Rs increased the number of seizures experienced, along with their severity. Rozmer and colleagues [32] hypothesised from these results that P2Y1 receptors increase proliferation and migration of NPCs, while P2X7R mediated necrosis and apoptosis may counter these effects, which would otherwise result in chronic recurrent epileptic seizures.
Experiments have been carried out to block the P2X7R in order to fully understand the role of this receptor in perinatal stress and subsequent brain injury. P2X7R is over-expressed in a neonatal mouse model of global hypoxia, and targeting of P2X7R with A-438079, a receptor antagonist of P2X7R, can reduce the number of post-hypoxia neonatal seizures[40]. These results corroborated an earlier study by Mesuret and colleagues [33], which used the same inhibitor to investigate the effects of P2X7R antagonism on early-life seizures in rats. This study also found that P2X7R blockade by A-438079 improved neonatal seizures, and suggested A-438079 could be used as a treatment for neonatal seizures or paediatric status epilepticus[33]. Similarly, Brilliant Blue G (BBG), a P2X7R-specific inhibitor, inhibits LPS-induced IL-1β release in mouse models of intrauterine inflammation [34], resulting in perinatal brain injury. P2X7R blockade resulted in reduced preterm birth rates, dendritic arborisation and density of cortical neurons, and improved performance for offspring in neuromotor tests [34]. These results supported the role of IL-1β as a key mediator of perinatal brain injury. Further studies corroborated the neuroprotective effects of P2X7R blockade, with da Silva and colleagues[35] showing that in a neonatal rat model of LPS-induced inflammation, pharmacologic blockade of P2X7R in the neonatal period using BBG has neuroprotective effects that persist into adulthood [35].
4.1. Cytokines and Chemokines in Neonatal Encephalopathy
Cytokines and chemokines, such as tumour necrosis factor α (TNFα) and interleukin 1β (IL1β), are released by microglia and astrocytes in response to hypoxic ischaemic injury, amplifying inflammatory cascades that recruit monocytes and neutrophils to the site of injury [36]. Studies have demonstrated an association between more adverse outcomes following perinatal brain damage and pro-inflammatory cytokines, such as TNFα, IL1β and IL6 [37]. These cytokines are released by astrocytes, neurons and microglia and are associated with HIE. A study by Liu and colleagues [37] demonstrated this by studying the peripheral blood levels of TNFα and IL1β of human neonates with HIE and control neonates. It was found that neonates with HIE consistently had higher levels of TNFα and IL1β, and there was a positive correlation between IL1β levels and HIE severity[37]. Chemokines also play a pivotal role in the inflammatory response following NE or HIE, due to their roles in inflammatory cell trafficking and leukocyte activation [38]. When HIE is modelled in rats [39], upregulation of alpha-chemokines such as macrophage inflammatory protein 2 (MIP2) and beta-chemokines such as MIP1α, MIP1β and CCL5 is induced, followed by the expression of lymphocyte markers in the site of infarction. This inflammatory response persisted beyond the neonatal period in this rat model, indicating that this acute inflammation may trigger a chronic inflammatory response[39].
4.1.1. Interleukin 1β, IL1β
There is no doubt that IL-1β levels are increased after neonatal brain injury. Hypoxic ischemia and hypoxia-only both cause acutely increased IL1β levels after the original insult[40][2]. However, it is not clear whether this increase of IL1β is sustained over time. Results from the hypoxia–ischemia model showed increases in IL1β 6 days and 14 days after reperfusion. However, in the hypoxia-only model, IL1 β levels returned to normal levels after 72 h[2].
Supporting the role of IL1 β in ischemia, administration of type 1 interleukin receptor (IL1R1) antagonist or blocking antibodies ameliorates damage induced by excitotoxicity and/or ischemia. IL1β knockdown by lentivirus in vivo can also improve the damage caused by neonatal HI[40]. Further analysis will be necessary if these results can be reproduced on the hypoxia-only model where the induction of IL1β is transient.
4.1.2. Interleukin-6, IL6
IL6 has been shown to have a dual function, having beneficial and/or detrimental effects depending on the context. In adult rodents, IL6 has an inflammatory effect in the acute phase, but during the chronic phase, IL6 acts as a neurotrophic factor[3].
In the neonatal brain, IL6 is increased transiently in the first hours after ischemia. Blockers of IL6 have a neuroprotective effect after the ischemic insult. However, no differences were observed between the ipsilateral (hypoxic-ischemic side) and the contralateral side (hypoxic only side), making it difficult to determine the role of IL6 in the neonatal brain. Further studies will be necessary to fully elucidate the role of IL6 during neonatal brain injury.
4.1.3. Tumour Necrosis Factor, TNFα
It is clear that TNFα is increased after neonatal brain injury; however, its role is not clear. TNFα binds to TNF-R1 and TNF-R2. Activation of TNF-R1 activates a caspase signalling pathway, resulting in cell death. In contrast, activation of TNF-R2 induces cell proliferation via the survival Akt signalling pathway. In neonatal hypoxic ischemia in rats, an increase of TNF-R1 has been observed in oligodendrocytes, suggesting that TNFα may play a role in the apoptosis and delayed myelination observed in neonatal brain injury. The role of TNFα will require further analysis to clarify its beneficial or detrimental effect [41].
References
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; et al. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208.
- Quinlan, S.; Merino-Serrais, P., Di Grande, A.; Dussmann, H.; Prehn, J.H.M.; Ní Chonghaile, T.; Henshall, D.C.; Jimenez-Mateos, E.M. The anti-inflammatory compound candesartan cilexetil improves neurological outcomes in a mouse model of neonatal hypoxia. Front Immunol. 2019. 10, 1752.
- Mallard, C.; Tremblay, M.E.; Vexler, Z.S. Microglia and Neonatal Brain Injury. Neuroscience 2019, 1, 68–76.
- Takemiya, T.; Fumizawa, K.; Yamagata, K.; Iwakura, Y.; Kawakami, M. Brain Interleukin-1 facilitates learning of a water maze spatial memory task in young mice. Front Behav. Neurosci. 2017, 11, 202.
- Al Mamun, A.; Yu, H.; Romana, S.; Liu, F. Inflammatory responses are sex specific in chronic hypoxic–ischemic encephalopathy. Cell Transplant. 2018, 1328–1339, doi:10.1177/0963689718766362.
- Manuck, T.A.; Rice, M.M.; Bailit, J.L.; et al. Preterm neonatal morbidity and mortality by gestational age: A contemporary cohort. Am. J. Obstet. Gynecol. 2016, 215, e101–e114.
- Schmidt, J.W.; Walsh, W.F. Hypoxic-ischemic encephalopathy in preterm infants. J. Neonatal. Perinatal. Med. 2010, 3, 277–284.
- Chalak, L.F.; Rollins, N.; Morriss, M.C.; Brion, L.P.; Heyne, R.; Sanchez, P.J. Perinatal acidosis and hypoxic-ischemic encephalopathy in preterm infants of 33 to 35 weeks’ gestation. J. Pediatr. 2012, 160, 388–394.
- Garfinkle, J.; Wintermark, P.; Shevell, M.I.; Oskoui, M. Children born at 32 to 35 weeks with birth asphyxia and later cerebral palsy are different from those born after 35 weeks. J. Perinatol. 2017, 37, 963–968.
- Woodward, L.J.; Anderson, P.J.; Austin, N.C.; Howard, K.; Inder, T.E. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 2006, 355, 685–694.
- Batalle, D.; Eixarch, E.; Figueras, F.; et al. Altered smallworld topology of structural brain networks in infants with intrauterine growth restriction and its association with later neurodevelopmental outcome. NeuroImage 2012, 60, 1352–1366.
- Mullen, K.M.; Vohr, B.R.; Katz, K.H.; Schneider, K.C.; Lacadie, C.; Hampson, M.; Makuch, R.W.; Reiss, A. L.; Constable, R.T.; Ment, L.R. Preterm birth results in alterations in neural connectivity at age 16 years. NeuroImage 2011, 54, 2563–2570.
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109.
- Bartha, A.I.; Foster-Barber, A.; Miller, S.P.; Vigneron, D.B.; Glidden, D.V.; Barkovich, A.J.; Ferriero, D.M Neonatal encephalopathy: Association of cytokines with MR spectroscopy and outcome. Pediatr. Res. 2004, 56, 960–966.
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518.
- Serdar, M.; Kempe, K.; Rizazad, M.; Herz, J.; Bendix, I.; Felderhoff-Müser, U.; Sabir, H. Early pro-inflammatory microglia activation after inflammation-sensitized hypoxic-ischemic brain injury in neonatal rats. Front Cell Neurosci. 2019, 13, 237, doi:10.3389/fncel.2019.00237.
- Lalancette-Hébert, M.; Faustino, J.; Sampath Thammisetty, S.; Chip, S.; Vexler, Z.S.; Kriz, J. Live imaging of the innate immune response in neonates reveals differential TLR2 dependent activation patterns in sterile inflammation and infection. Brain Behav. Immun. 2017, 65, 312–327.
- Stridh, L.; Smith, P.L.; Naylor, A.S.; Wang, X; Mallard, C. Regulation of Toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. J. Neuroinflammation 2011. 8, 45.
- Mottahedin, A.; Ardalan, M.; Chumak, T.; Riebe, I.; Ek, J.; Mallard, C. Effect of neuroinflammation on synaptic organization and function in the developing brain: Implications for neurodevelopmental and neurodegenerative disorders. Front. Cell. Neurosci. 2017, 11, 190.
- Tang, Z.; Cheng, S.; Sun, Y.; Zhang, Y.; Xiang, X.; Ouyang, Z.; Zhu, X.; Wang, B.; Hei, M. Early TLR4 inhibition reduces hippocampal injury at puberty in a rat model of neonatal hypoxic-ischemic brain damage via regulation of neuroimmunity and synaptic plasticity. Exp. Neurol. 2019, 321, 113039.
- Burnstock, G. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug. Disc. 2008. 7, 575–590.
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797, doi:10.1152/physrev.00043.2006.
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trend Pharmacol Sci 2014, 35, 537–547.
- Rappold, P.M.; Lynd-Balta, E.; Joseph, S.A. P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 2006, 1089,171–178.
- Mingam, R.; De Smedt, V.; Amedee, T.; Bluthe, R.M.; Kelley, W.K.; Dantzer, R.; Laye, S. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1 beta in the murine brain. Brain Behav. Immun. 2008, 22, 234–244.
- Monif, M.; Reid, C.A.; Powell, K.L.; Smart, M.L.; Williams, D.A. The P2X7 receptor drives microglial activation and proliferation: A trophic role for P2X7R pore. J. Neurosci. 2009, 29, 3781–3791.
- Armstrong, J.N.; Brust, T.B.; Lewis, R.G.; MacVicar, B.A. Activation of presynaptic P2X7-like receptors depresses mossy fiber-CA3 synaptic transmission through p38 mitogen-activated protein kinase. J. Neurosci. 2002, 22, 5938–5945.
- Sperlagh, B.; Köfalvi, A.; Deuchars, J.; Atkinson, L.; Milligan, C.J.; Buckley, N.J.; Vizi, E.S. Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J. Neurochem. 2002, 81, 1196–1211.
- Dale, N.; Frenguelli, B.G. Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 2009, 7, 160–179.
- Ulrich, H.; Illes, P. P2X receptors in maintenance and differentiation of neural progenitor cells. Neural. Regen. Res. 2014, 9, 2040–2041.
- Illes, P.; Messemer, N.; Rubini, P. P2Y receptors in neurogenesis. WIRES Membr. Transp. Signal 2013, 2, 43–48.
- Rozmer, K.; Gao, P.; Araújo, M.G.L.; Tahir Khan, M.; Liu, J.; Rong, W.; Tang, Y.; Franke, H.; Krügel, U.; Fernandes, M.J.S.; Illes, P. Pilocarpine-Induced Status Epilepticus Increases the Sensitivity of P2X7 and P2Y1 Receptors to Nucleotides at Neural Progenitor Cells of the Juvenile Rodent Hippocampus. Cereb. Cortex 2017, 27, 3568–3585.
- Mesuret, G.; Engel, T.; Hessel, E.V.; Sanz-Rodriguez, A.; Jimenez-Pacheco, A.; Miras-Portugal, M.T.; Diaz-Hernandez, M.; Henshall, D.C. P2X7 receptor inhibition interrupts the progression of seizures in immature rats and reduces hippocampal damage. CNS Neurosci. Ther. 2014, 20, 556–564, doi: 10.1111/cns.12272.
- Tsimis, M.E.; Lei, J.; Rosenzweig, J.M.; Arif, H.; Shabi, Y.; Alshehri, W.; Talbot, C.C.; Baig-Ward, K.M.; Segars, J.; Graham, E.M.; Burd, I. P2X7 receptor blockade prevents preterm birth and perinatal brain injury in a mouse model of intrauterine inflammation. Biol. Reprod. 2017, 97, 230–239.
- Da Silva, C.S.; Longoni Calió, M.; Mosini, A.C.; Moreira Pires, J.; da Silva Bandeira Rêgo, D.; Mello, L.E.; Figueiredo Stochero Leslie, A.T. LPS-Induced systemic neonatal inflammation: Blockage of P2X7R by BBG decreases mortality on rat pups and oxidative stress in hippocampus of adult rats. Front. Behav. Neurosci. 2019, 13, 240.
- Jenkins, D.D.; Rollins, L.G.; Perkel, J.K.; Wagner, C.L.; Katikaneni, L.P.; Bass, W.T.; Kaufman, D.A.; Horgan, M.J.; Languani, S.; Givelichian, L.; et al. Serum cytokines in a clinical trial of hypothermia for neonatal hypoxic-ischemic encephalopathy. J. Cereb. Blood Flow Metab. 2012, 32, 1888–1896, doi: 10.1038/jcbfm.2012.83.
- Liu, J.; Feng, Z.C. Increased umbilical cord plasma interleukin-1 beta levels was correlated with adverse outcomes of neonatal hypoxic-ischemic encephalopathy. J. Trop. Pediatr. 2010, 56, 178–182.
- Liu, F.; Mccullough, L.D. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol. Sin. 2013, 34, 1121–1130.
- Bona, E.; Andersson, A.L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; et al. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res. 1999, 45, 500–509.
- Rodriguez-Alvarez, N.; Jimenez-Mateos, E.M.; Engel T.; Quinlan, S.; Reschke, C.R.; Conroy R.M.; Bhattacharya, A.; Boylan, G.B.; Henshall, D.C. Effects of P2X7 receptor antagonists on hypoxia-induced neonatal seizures in mice. Neuropharmacology 2017, 116, 351–363.
- Winerdal, M.; Winerdal, M.E.; Kinn, J.; Urmaliya, V.; Winqvist, O.; Ådén, U. Long lasting local and systemic inflammation after cerebral hypoxic ischemia in newborn mice. PLoS ONE 2012, 7, e36422.

