 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paul Quax | + 2090 word(s) | 2090 | 2021-01-15 05:24:31 | | | |
| 2 | Vicky Zhou | Meta information modification | 2090 | 2021-01-18 07:17:29 | | |
Video Upload Options
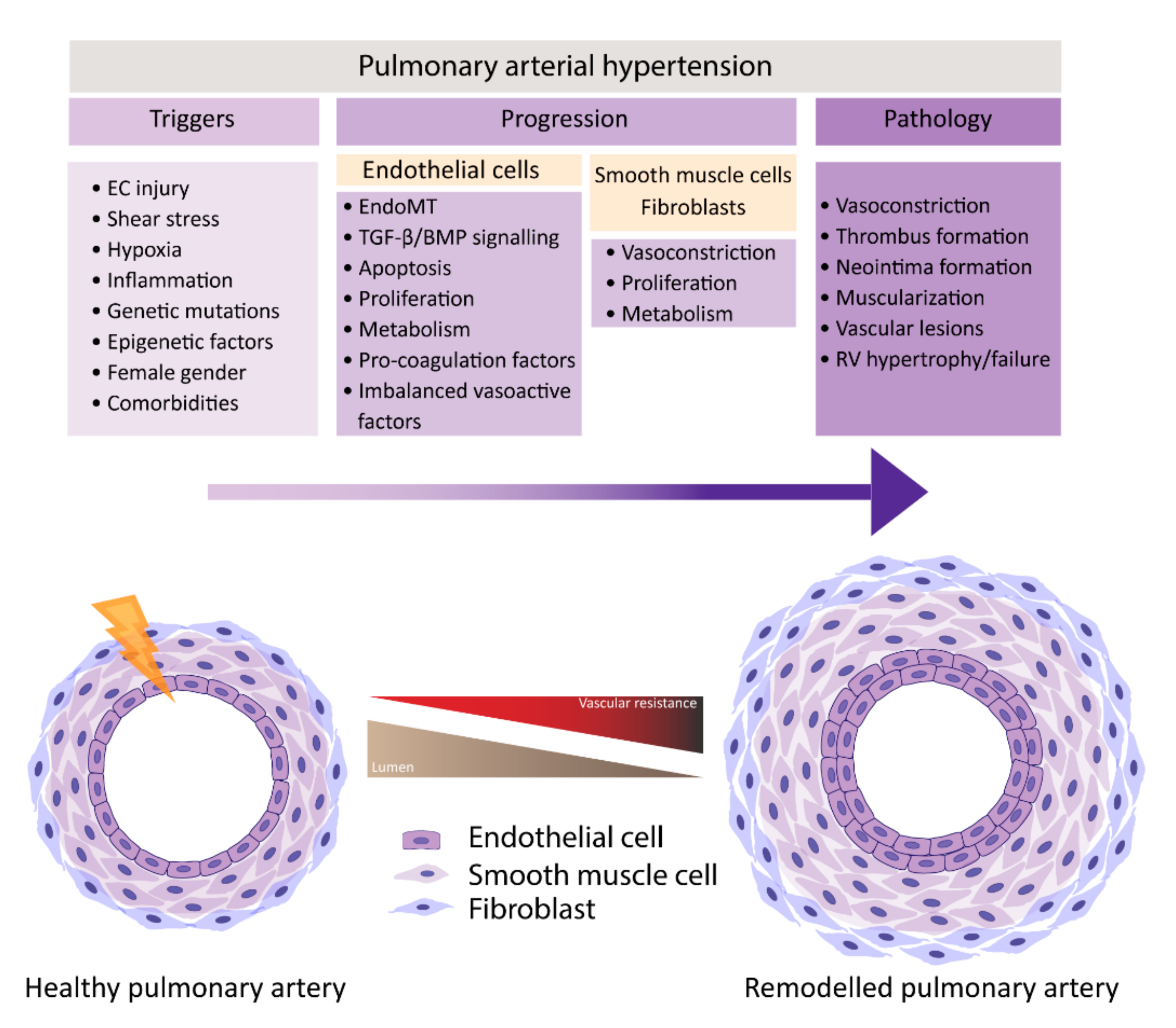
Endothelial Cell (EC) dysfunction is one of the first triggers initiating the process of Pulmonary Arterial Hypertension (PAH). EC dysfunction leads to the activation of several cellular signalling pathways in the endothelium, resulting in the uncontrolled proliferation of ECs, pulmonary artery smooth muscle cells, and fibroblasts, and eventually leads to vascular remodelling and the occlusion of the pulmonary blood vessels. Other factors that are related to EC dysfunction in PAH are an increase in endothelial to mesenchymal transition, inflammation, apoptosis, and thrombus formation.
1. Introduction
Pulmonary hypertension (PH) is a condition that is defined by a mean pulmonary arterial pressure of more than 20 mmHg at rest and 30 mmHg during exercise. The range of genetic, molecular, and humoral causes that can lead to this increase in pressure is extensive. Therefore, PH is grouped into different classes that are based on clinical and pathological findings as well as therapeutic interventions [1][2]. The World Health Organization (WHO) classifies PH into five groups, namely: 1. Pulmonary arterial hypertension (PAH), 2. Pulmonary hypertension due to left heart disease (PH-LHD), 3. Pulmonary hypertension due to lung disease (PH-LD), 4. Chronic thromboembolic pulmonary hypertension (CTEPH), and 5. Pulmonary hypertension due to unclear and/or multifactorial mechanisms [1][3][4]. PH is increasingly becoming a global health issue due to the ageing population. Although PH-LHD and PH-LD are the most prevalent PH groups, research and drug development mainly focus on PAH and CTEPH, which are rarer diseases that mainly affect younger people [5]. This review will focus mostly on PAH because of the amount of research conducted in PAH as compared to the other four groups.
PAH is characterized by remodeling of distal pulmonary arteries, causing a progressive increase in vascular resistance. Vascular remodeling is associated with alterations in vasoconstriction, pulmonary artery- endothelial cells (PAECs) and -smooth muscle cells (PASMCs) cell proliferation, inflammation, apoptosis, angiogenesis, and thrombosis, which leads to the muscularization and occlusion of the lumen of pulmonary arteries by the formation of vascular lesions. Some of the lesions found in PAH are plexiform lesions, which are characterized by enhanced endothelial cell (EC) proliferation, thrombotic lesions and neointima formation, the formation of a layer of myofibroblasts, and extracellular matrix between the endothelium and the external elastic lamina [6][7]. One of the first triggers for development of PAH is thought to be EC injury triggering the activation of cellular signaling pathways that are not yet completely understood.
In normal conditions, the endothelium is in a quiescent and genetically stable state. When activated, the endothelium secretes different growth factors and cytokines that affect EC and SMC proliferation, apoptosis, coagulation, attract inflammatory cells, and/or affect vasoactivity in order to restore homeostasis. Prolonged or chronic activation of the endothelium leads to EC dysfunction, the loss of homeostatic functions, leading to pathological changes, and it is crucial in the development of cardiovascular diseases and so too in PAH [8][9]. Many different factors have been suggested to be triggers of EC dysfunction in PAH, like shear stress, hypoxia, inflammation, cilia length, and genetic factors (Figure 1) [6][10][11][12]. As a consequence, the endothelium switches from a quiescent to an overactive state, where it starts to secrete vasoconstrictive factors, like endothelin-1 (ET-1) [13] and thromboxane [14], and proliferative factors, like vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2) [15], CXCL12 [16], and reduce the secretion of vasodilators, like nitric oxide (NO) and prostacyclin, which indicates that EC dysfunction might play a central role in the pathogenesis of PAH. Whether EC dysfunction is the primary cause or rather the consequence of changes in environmental factors remains to be resolved [8][17].
2. Features of EC Dysfunction
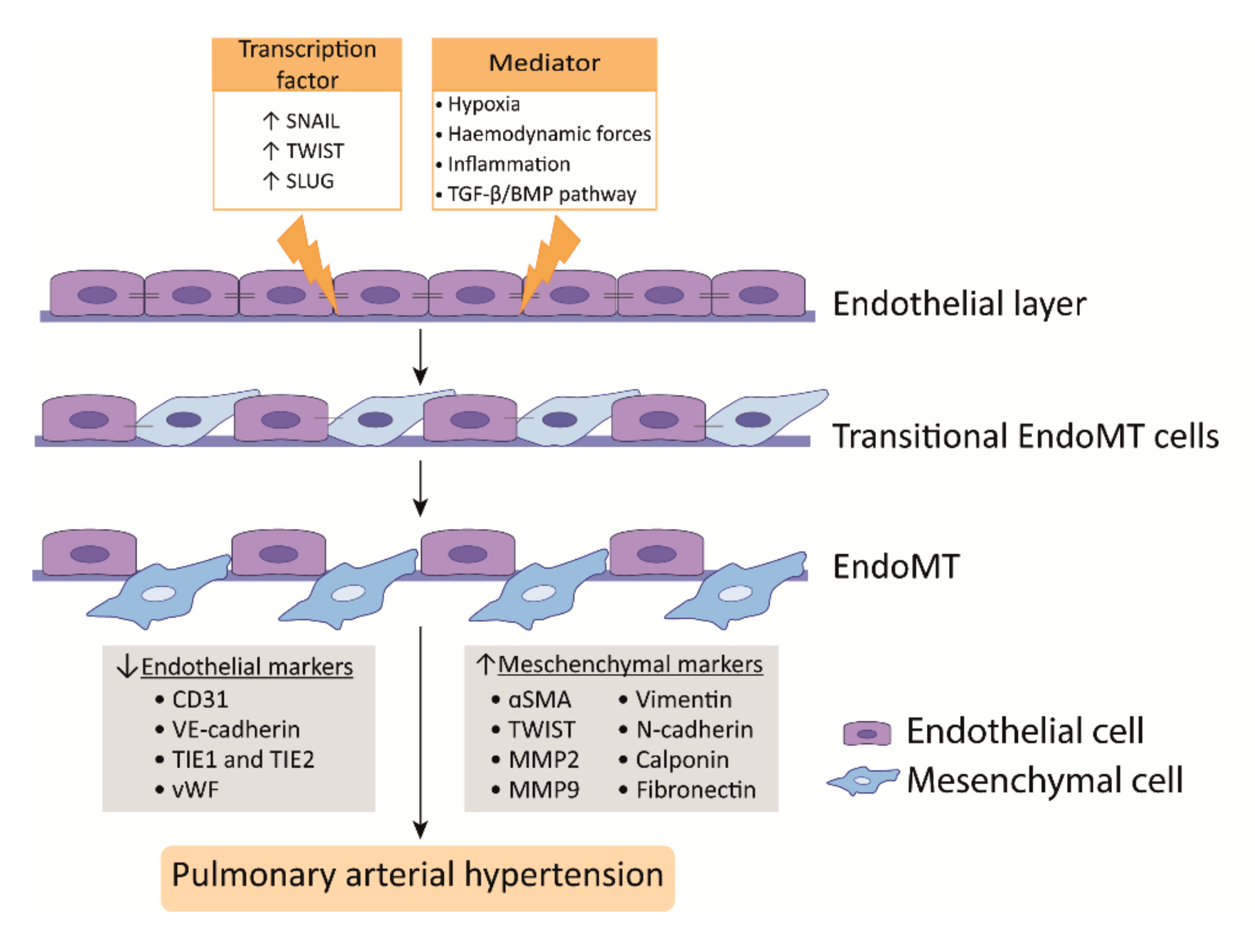
PAH is characterized by a dysfunctional endothelium, of which the balance between vasodilation and vasoconstriction, but also the growth factor production and cell survival are altered. In addition, ECs undergo endothelial to mesenchymal transition (EndoMT), which, all together, causes perturbations in pulmonary vascular homeostasis that promote vascular remodeling (Figure 2).
2.1. Perturbations in Vasoactivity
Reduced vasorelaxation in PAH mainly contributes to the altered expression of the vasodilators NO and prostacyclin. NO is a fast-reacting endogenous free radical that is produced by endothelial NO Synthase (eNOS). NO is essential for vasorelaxation via PASMCs, but it also has antithrombotic effects and controls EC differentiation and growth [18][19][20]. NO has long been implicated in the pathogenesis of PAH, and the lungs of PAH patients have reduced NO expression [21]. Whole exome sequencing has identified that mutations in Caveolin-1 are associated with PAH. Caveolin-1 is highly expressed in ECs and, interestingly, the C-terminus of caveolin-1 directly interacts with eNOS, which may result in the disruption in NO levels, ultimately triggering PAH [22]. However, other studies reported contradictory results and some PH patients even show an increase in eNOS expression [21]. Furthermore, eNOS-/- mice show reduced vascular remodeling after chronic hypoxia that is caused by reduced vascular proliferation [23], pointing out the complexity of its role in PAH. Prostacyclin, also produced by EC with additional antithrombotic and antiproliferative properties [8][24][25][26], is synthesized from arachidonic acid, by prostacyclin synthase, and cyclo-oxygenase (COX) [27]. Decreased prostacyclin levels are measured in various patients with different forms of PAH, like iPAH and HIV-associated PAH [8][28], explaining, in part, the increase in pulmonary vasoconstriction, SMC proliferation, and coagulation occurring in these patients. Interestingly, in experimental PH models, mice overexpressing prostacyclin synthase are protected from developing chronic hypoxia-induced PAH [29].
ET-1, on the other hand, is a potent vasoconstrictor, which is mainly synthesized in EC and the lungs show the highest level of ET-1 in the entire body [30]. ET-1 exhibits its effects by binding to the ETA and ETB receptors, which activate signalling pathways in vSMCs regulating proliferation, vasorelaxation and vasoconstriction [26][30]. ETA is predominantly expressed on vSMCs and is involved in vasoconstriction and proliferation of these cells, while ETB is expressed on vSMCs and PAECs, and is involved in stimulating the release of vasodilators, like NO and prostacyclin, and the inhibition of apoptosis [31][26][30][32][33]. The expression of ET-1 and its receptors is increased in lungs of PAH patients and experimental PH models [34][35][36][37]. Furthermore, a correlation exists between the expression of ET-1 and an increase in pulmonary resistance in PAH [36]. The increased synthesis of endothelial ET-1, accompanied with an increase in expression of ETA on PASMCs, likely contributes to the increased vasoconstriction and vascular remodelling observed in PAH [25][37][38]. Another vasoconstrictor, thromboxane A2, which is produced by ECs and platelets, but is also an inducer of platelet aggregation and a vSMCs mitogen, is increased in PAH [8][14], creating an imbalance that might contribute to excessive platelet aggregation and vascular remodeling observed in PAH [14].
At last, the expression of the growth factor vascular endothelial growth factor (VEGF) and its receptor VEGF receptor 2 (VEGFR2) are found to be increased in ECs from plexiform lesions from iPAH patients. Additionally, the plasma levels of VEGF are found to be elevated in PH patients [39][40]. The relation between PAH and increased VEGF expression is still poorly understood. It is suggested that VEGF levels in PAECs are elevated in early stages of PAH as a protective response, while, during disease progression, VEGF keeps promoting the growth of PAECs, causing the formation of plexiform lesions [8].
2.2. Endothelial to Mesenchymal Transition
EndoMT is a phenomenon where ECs acquire a mesenchymal-like phenotype that is accompanied with a loss of endothelial markers and increase of mesenchymal markers. In addition, ECs lose cell-cell contact, change their morphology, and adopt a highly migratory and invasive phenotype, thereby losing features of a healthy endothelium (Figure 2) [41][42]. In the lungs of human PAH patients and monocrotaline (MCT) and Sugen/hypoxia (SuHx) experimental PH rat models, EndoMT was observed, whereby cells express high levels of α-SMA and activated phospho-vimentin and VE-cadherin, indicating their endothelial origin [43][44][45]. Moreover, TWIST1, which is a key transcription factor in inducing EndoMT, is highly expressed in human PAH lungs as compared to healthy lungs [44] (Figure 2).
TGFβ treatment of PAECs induces the expression of the EndoMT transcription factors TWIST1 and SNAIL1 [41][46] and the mesenchymal markers α-SMA and phospho-vimentin [47]] (Figure 2). TWIST1 increases the expression of TGFβ, leading to enhanced TGFβ signaling [48]. In addition, reduced BMPR2 signaling promotes EndoMT via the upregulation of the High Mobility Group AT-hook 1 and its target gene SLUG, independent of TGFβ signaling [49]. More interestingly, BMP-7, a protein previously described as having anti-inflammatory and anti-tumor effects in several diseases, was attenuated by hypoxia-induced EndoMT in PAECs both in vivo and in vitro by inhibiting the m-TORC1 signaling pathway [50]. BMPR2 loss favors EndoMT, allowing for cells of myo-fibroblastic character to create a vicious feed-forward process, leading to hyperactivated TGFβ signaling [51]. In summary, alterations in TGFβ/BMP signaling are linked to the process of EndoMT that was observed in PAH [52].
Hypoxia is also an inducer of EndoMT through hypoxia-inducible transcription factor-1α (HIF-1α) and HIF-2α, and both transcription factors are increased in PAH [53][54] (Figure 2). PAH ECs display an increased expression of HIF-2α, leading to SNAIL upregulation [45]. In addition, HIF-1α knockdown alone effectively blocks hypoxia-induced EndoMT, but also the knockdown of its downstream target gene TWIST1 showed the effective blockage of hypoxia-induced EndoMT in microvascular ECs (MVECs); however, it was less pronounced [55]. Nonetheless, it is important to realize that microvascular endothelium may differ from arterial endothelial function. Finally, in addition to transcription factors, microRNAs, such as miR-181b, have been shown to be implicated in EndoMT in PAH. The overexpression of miR-181b in rat pulmonary arterial ECs (rPAECs) attenuated inflammation-induced EndoMT by inhibiting the expression of TGF-βR1 and circulating proteoglycan endocan [56].
2.3. Apoptosis
EC apoptosis may also play a role in PH development via vascular dropout and selection pressure on ECs, contributing to the apoptosis-resistant phenotype of ECs in vascular lesions [57]. Several attempts were made in order to elucidate the molecular pathways that are involved in the regulation of PAEC apoptosis. The hypothesis is that disturbed responses to VEGF signaling, in combination with hypoxia, cause an initial increase in apoptosis in PAECs, leading to the emergence of aggressive apoptosis resistant and hyperproliferative ECs that cause the formation of intimal lesions [58][59][60]. A possible explanation for the initial increase in apoptosis of PAECs is that the loss of BMPR2 signaling promotes mitochondrial dysfunction and subsequent PAEC apoptosis [61]. White et al., interestingly, proposes a model in which the pro-apoptotic factor programmed cell death-4 (PDCD4) activates the cleavage of caspase-3, inducing PAEC apoptosis. Interestingly, they show that reducing PDCD4 levels in vivo by overexpressing miRNA-21 prevents PH development in SuHx rats [62]. Besides an initial increase in apoptosis, PAH is also characterized by PAECs that are hyperproliferative and apoptosis resistant [60]. PAECs from iPAH patients showed an increased expression of pro-survival factors IL-15, BCL-2, and Mcl-1, together with persistent activation of the pro-survival STAT3 signaling pathway [60]. Furthermore, Notch1 was elevated in lungs from iPAH patients and from SuHx rats. Notch1 contributes to PAH pathogenesis by increasing EC proliferation and inhibiting apoptosis via p21 downregulation and regulating BCL-2 and survivin expression. Furthermore, HIF1α expression promotes Notch signaling human PAECs [63]. In contrast, Miyagawa et al., demonstrated that contact-mediated communication between SMC and EC activates EC derived Notch1 and alters the cells epigenome in order to regulate Notch1-dependent genes that maintain endothelial integrity and prevent pulmonary vascular remodeling in a murine model of hypoxia-induced pulmonary hypertension [64]. Therefore, the role of Notch1 is complex and controversial in PAH and warrants more research to delineate the molecular mechanisms.
References
- Dumitrescu, D.; Hager, A.; Held, M.; Sinning, C.; Greiner, S.; Kruck, I.; Meyer, J.; Pabst, S.; Köhler, T.; Kovacs, G.; et al. Definition, clinical classification and initial diagnosis of pulmonary hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 11–19.
- Simonneau, G.; Hoeper, M.M. The revised definition of pulmonary hypertension: Exploring the impact on patient management. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2019, 21, K4–K8.
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913.
- Vonk Noordegraaf, A.; Groeneveldt, J.A.; Bogaard, H.J. Pulmonary hypertension. Eur. Respir. Rev. 2016, 25, 4.
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.C.; Gibbs, J.S.R. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322.
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S.
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285.
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial Dysfunction in Pulmonary Hypertension. Circulation 2004, 109, 159–165.
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198.
- Humbert, M.; Montani, D.; Perros, F.; Dorfmüller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vasc. Pharmacol. 2008, 49, 113–118.
- Nicod, L.P. The endothelium and genetics in pulmonary arterial hypertension. Swiss Med. Wkly. 2007, 137, 437–442.
- Dummer, A.; Rol, N.; Szulcek, R.; Kurakula, K.; Pan, X.; Visser, B.I.; Bogaard, H.J.; DeRuiter, M.C.; Goumans, M.J.; Hierck, B.P. Endothelial dysfunction in pulmonary arterial hypertension: Loss of cilia length regulation upon cytokine stimulation. Pulm. Circ. 2018, 8.
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469.
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 70–75.
- Tu, L.; Dewachter, L.; Gore, B.; Fadel, E.; Dartevelle, P.; Simonneau, G.; Humbert, M.; Eddahibi, S.; Guignabert, C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 311–322.
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802.
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.J.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.A.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis In Vivo and endothelial cell growth and migration In Vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044.
- Babaei, S.; Teichert-Kuliszewska, K.; Monge, J.C.; Mohamed, F.; Bendeck, M.P.; Stewart, D.J. Role of nitric oxide in the angiogenic response In Vitro to basic fibroblast growth factor. Circ. Res. 1998, 82, 1007–1015.
- Giaid, A.; Saleh, D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1995, 333, 214–221.
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343.
- Quinlan, T.R.; Li, D.; Laubach, V.E.; Shesely, E.G.; Zhou, N.; Johns, R.A. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000.
- Epstein, F.H.; Vane, J.R.; Änggård, E.E.; Botting, R.M. Regulatory Functions of the Vascular Endothelium. N. Engl. J. Med. 2010, 323, 27–36.
- Chen, Y.F.; Oparil, S. Endothelial Dysfunction in the Pulmonary Vascular Bed. Am. J. Med. Sci. 2000, 320, 223–232.
- Humbert, M.; Sitbon, O. Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2004, 351, 1425–1436.
- Mitchell, J.A.; Ahmetaj-Shala, B.; Kirkby, N.S.; Wright, W.R.; Mackenzie, L.S.; Reed, D.M.; Mohamed, N. Role of prostacyclin in pulmonary hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 382–393.
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999.
- Geraci, M.W.; Gao, B.; Shepherd, D.C.; Moore, M.D.; Westcott, J.Y.; Fagan, K.A.; Alger, L.A.; Tuder, R.M.; Voelkel, N.F. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J. Clin. Investig. 1999, 103, 1509–1515.
- Chester, A.H.; Yacoub, M.H. The role of endothelin-1 in pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 62–78.
- Veyssier-Belot, C.; Cacoub, P. Role of endothelial and smooth muscle cells in the physiopathology and treatment management of pulmonary hypertension. Cardiovasc. Res. 1999, 44, 274–282.
- Shao, D.; Park, J.E.S.; Wort, S.J. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharm. Res. 2011, 63, 504–511.
- Shichiri, M.; Kato, H.; Marumo, F.; Hirata, Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 1997, 30, 1198–1203.
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739.
- Li, H.B.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced Endothelin-1 and Endothelin Receptor Gene-Expression in Chronic Hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459.
- Frasch, H.F.; Marshall, C.; Marshall, B.E. Endothelin-1 is elevated in monocrotaline pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L304–L310.
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405.
- Galié, N.; Manes, A.; Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 2004, 61, 227–237.
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374.
- Säleby, J.; Bouzina, H.; Ahmed, S.; Lundgren, J.; Rådegran, G. Plasma receptor tyrosine kinase RET in pulmonary arterial hypertension diagnosis and differentiation. ERJ Open Res. 2019, 5, 00037–02019.
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508.
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384.
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018.
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858.
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α Contributes to Severe Pulmonary Hypertension by Inducing Endothelial-to-Mesenchymal Transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 314, L256–L275.
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298.
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thomé, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faça, V.; Olsen Câmara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-β2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458.
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-β–Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207.
- Hopper, R.K.; Moonen, J.R.A.J.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In pulmonary arterial hypertension, reduced bmpr2 promotes endothelial-to-Mesenchymal transition via hmga1 and its target slug. Circulation 2016, 133, 1783–1794.
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell. Physiol. 2018.
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557.
- Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585.
- Lei, W.; He, Y.; Shui, X.; Li, G.; Yan, G.; Zhang, Y.; Huang, S.; Chen, C.; Ding, Y. Expression and analyses of the HIF-1 pathway in the lungs of humans with pulmonary arterial hypertension. Mol. Med. Rep. 2016, 14, 4383–4390.
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434.
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelial-mesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278.
- Zhao, H.; Wang, Y.; Zhang, X.; Guo, Y.; Wang, X. miR-181b-5p inhibits endothelial-mesenchymal transition in monocrotaline-induced pulmonary arterial hypertension by targeting endocan and TGFBR1. Toxicol. Appl. Pharmacol. 2020, 386, 114827.
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12.
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438.
- Sakao, S.; Taraseviciene-Stewart, L.; Lee, J.D.; Wood, K.; Cool, C.D.; Voelkel, N.F. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1178–1180.
- Masri, F.A.; Xu, W.; Comhair, S.A.; Asosingh, K.; Koo, M.; Vasanji, A.; Drazba, J.; Anand-Apte, B.; Erzurum, S.C. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, 548–554.
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608.
- White, K.; Dempsie, Y.; Caruso, P.; Wallace, E.; McDonald, R.A.; Stevens, H.; Hatley, M.E.; Van Rooij, E.; Morrell, N.W.; Maclean, M.R.; et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014, 64, 185–194.
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149.
- Miyagawa, K.; Shi, M.; Chen, P.I.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S.; et al. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ. Res. 2019, 124, 211–224.

