 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Timo Gaber | + 2823 word(s) | 2823 | 2021-01-11 04:04:21 | | | |
| 2 | Camila Xu | Meta information modification | 2823 | 2021-01-14 09:14:38 | | | | |
| 3 | Camila Xu | Meta information modification | 2823 | 2021-01-14 09:21:07 | | |
Video Upload Options
The Janus kinase (JAK) signal transducer and activator of transcription (STAT) signaling pathway serves as an important downstream mediator for a variety of cytokines, hormones, and growth factors. Based on the knowledge gained from JAK and STAT knockout animals, the JAK/STAT signaling pathway was identified as important for bone development and homeostasis, recognizing that JAKs and STATs are not equally important for the biology of osteoblasts and osteoclasts. Moreover, their overall role in the musculoskeletal system is still not fully understood. Understanding the underlying mechanisms of how bone remodeling is regulated, how metabolic processes take place, and how bone responds to mechanical stimulation is central to maintaining the integrity of the skeletal system.
1. Introduction
The sense of and reaction to external signals from the environment is essential for the survival of every living system. At the level of the whole organism, the sensory organs such as eyes, ears, and skin are specialized in perceiving the signals of the environment, processing the incoming signals, and passing on the information to finally trigger a reaction of the whole body. At the cellular level, external signals are primarily sensed and processed by biochemical receptors in the cell membrane and transmitted via signaling pathways and cascades that form a network with a variety of other pathways to further process the information. These signals initiate mechanisms that are responsible for controlling phenotypic and functional outcomes, e.g., proliferation or apoptosis. Among these signal transduction pathways, the Janus tyrosine kinase (JAK)- and signal transducers and activators of transcription (STAT)-mediated signaling are responsible for transducing signals of more than fifty cytokines, growth factors and hormones, regulated on multiple levels [1][2][3]. Loss- or gain-of-function mutations of genes encoding JAK/STAT components display dramatic immunological phenotypes in humans and mice underpinning the importance of the central communication hub for the immune system [1][3][4]. Regulation of cellular, molecular, and genomic processes via JAK and/or STAT proteins are inhibited by the suppressor of cytokine signaling (SOCS)—a family of intracellular negative feedback proteins (Figure 1). Some of these cytokines, growth factors, and hormones have been shown to regulate bone homeostasis via JAK and/or STAT proteins [5].
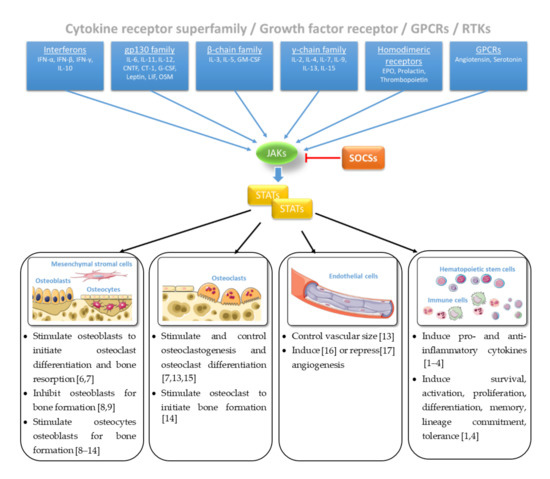
Figure 1. Janus tyrosine kinase (JAK)/signal transducers and activators of transcription (STAT) signaling in bone homeostasis [1][2][3][4][6][7][8][9][10][11][12][13][14][15][16][17]. Figure contains graphics from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
2. JAK/STAT Pathway at a Glance
In mammals, the JAK family contains four members (JAK1, JAK2, JAK3, and tyrosine kinase 2; TYK2). Their clinical importance has been highlighted by a human immunodeficiency syndrome caused by loss-of-function mutations in JAK3 [18][19]. Extracellular interaction of a cytokine with its transmembrane receptor initiates the canonical JAK/STAT signaling by inducing receptor oligomerization and trans-activation of JAKs. In turn, JAK trans-activation phosphorylates the cytoplasmatic domains of the receptor, which assist as docking sites for STATs. Spatial proximity of JAK and STAT facilitates JAK-mediated tyrosine-phosphorylation of STAT that dimerizes and translocates to the nucleus. In the nucleus, all phosphorylated STAT dimers bind to interferon-γ (IFN-γ)-activated sequence (GAS) DNA motifs except STAT2, which forms a trimeric complex with STAT1 and Interferon Regulatory Factor 9 (IRF9). Finally, the STAT1–STAT2–IRF9 complex also known as Interferon-stimulated Gene Factor 3 (ISGF3) engages the Interferon-stimulated Response Element (ISRE) motif (Figure 2). While JAK1, JAK2, and TYK2 are ubiquitously expressed, JAK3 is expressed more restricted, regulated, and tissue specific and can be found in hematopoietic cells such as NK cells, thymocytes, T cells, B cells, and myeloid cells but also in vascular smooth muscle cells and endothelium [2]. The name of the Janus kinases is based on the depiction of the Roman gate and door god “Janus” with his two faces and is based on their two-sided character featured by the existence of tandem kinase and pseudokinase domains [2]. Seven JAK homology (JH) regions are described. While the catalytic JH1 domain or kinase domain, which has all the characteristics of a typical tyrosine kinase domain, is well described, the function of the other JH regions is still poorly understood [2]. The JH2 domain is a so called pseudokinase domain that contains all of the subdomains that correspond to those in the catalytic JH1 tyrosine kinase but being altered from the typical subdomain motifs. The exact function remains to be elusive although being important for full functionality of the kinase domain and providing a docking site that associates with STATs. Both, the JH2-like domain and the FERM domain facilitate the interaction between JAKs and multiple upstream receptors [20][21][22][23].
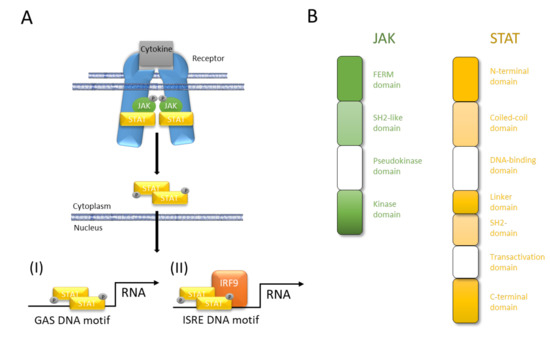
Figure 2. JAK/STAT pathway at a glance. (A) Cytokines interact with their corresponding receptor, which, after oligomerization, activates JAK and initiates JAK-mediated phosphorylation of its own cytoplasmic domain. Receptor phosphorylation causes STAT binding in close proximity to JAK that in turn mediates tyrosine-phosphorylation (p-Tyr) of the latter. STAT phosphorylation results in dimerization, nuclear translocation, DNA binding, and modulation of gene transcription. (I) All STAT can bind to interferon-γ (IFN-γ)-activated sequence (GAS) DNA motifs while (II) only STAT2 after forming a trimeric complex of STAT1–STAT2–IRF9 engages Interferon-stimulated Response Element (ISRE) DNA binding. (B) Four domains of JAK facilitate interaction with upstream receptors and promotion of kinase function (FERM domain), interaction with upstream receptors (SH2-like domain), control of kinase activity (pseudokinase domain), and trans-activation and tyrosine-phosphorylation of receptors, JAKs and STATs (kinase domain). The seven domains of STAT facilitate protein-protein interactions (N-terminal domain), protein–protein interactions and nuclear-localization (coiled-coil domain), nuclear import, DNA binding, and transcriptional activity (DNA-binding domain), structural organization and transcriptional activity (linker domain), dimerization and interaction with upstream receptors (SH2 domain), canonical signaling (transactivation domain), canonical and non-canonical functions (C-terminal domain).
The STAT family is composed of seven members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6), which share seven characteristic protein domains [2]. These domains interact with the upstream receptors, with each other (i.e., dimerization and tetramerization) and with certain DNA motifs. STATs mainly act as transcription factors that directly bind to DNA regulatory elements and control the transcription of associated genes. STAT binding can be observed proximal to DNA responsive elements but also distal and far from protein-encoding genes [2]. These sites can be distinguished in majority as enhancers, epigenetic hotspots, and non-coding loci. Thus, it is noteworthy, that STATs bear the capability to bind DNA, to act as transcription factor, and to modify the epigenome; the latter by either controlling the expression of various chromatin modifiers, or by physical interactions between e.g., STATs and CBP/p300, which mediates histone acetylation [2]. However, all members of the STAT family are capable to directly bind to GAS elements but do also often bind to STAT-binding sites which do not contain GAS motifs or STATs physically interact with other transcriptional regulators without DNA binding. Moreover, different STATs tend to co-localize extensively as exemplified in the interleukin (IL)-2Rα gene locus [2]. All the different modes of action and the various combinations of JAK and STAT proteins make an investigation on their targets almost impossible.
3. Guiding Bone Development by Combining JAKs and STATs
The skeletal system, one of the most important systems of the human body, serves as the structural support center of the body, provides a framework for the attachment of tissues, protects vital organs, and helps to direct the forces necessary for movement. The physiological bone development processes that lead to the structure, strength, and size of the bone are controlled by several pathways. These pathways regulate cellular functions within the skeletal system, which consists of bone-forming cells (osteoblasts), resident cells that form the regulatory network (osteocytes), and bone-resorbing cells (osteoclasts). During bone formation and remodeling processes, osteoblasts, osteocytes, osteoclasts, and chondrocytes are markedly influenced by various cytokines and their receptors such as the IL-6 receptor that is characterized by tyrosine kinases of the JAK family. Of note, many bone-related cytokines involved in bone development have been described, including those that signal through JAK and STAT pathways such as the IL-6 family of cytokines [3]. In bone, IL-6 family cytokines such as IL-6, IL-11, oncostatin M (OSM), cardiotrophin 1 (CT-1), leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF) act via the gp130 (glycoprotein 130) that activates gp130-associated JAKs [5][8]. This IL-6 receptor subunit has been demonstrated to be essential for the normal skeletal development, to stimulate bone formation of osteoblasts and to primarily act through STAT3 signaling. STAT3-dependent cytokines also suppress gene products that inhibit osteoblast differentiation, such as sclerostin [5]. Furthermore, the importance of the JAK/STAT signaling pathway for bone development is also highlighted by their involvement in mechanotransduction. Kido et al. showed that mechanical unloading suppresses, and reloading enhances the IL11 expression in bone cells [24]. IL-11 has been shown to induce receptor activator of nuclear factor κB ligand (RANKL) expression and stimulate bone resorption in vivo [25]. Moreover, the epidermal growth factor receptor (EGFR) and its ligands strongly inhibit osteoblast differentiation and mineralization, as determined by the decreased expression of the transcription factor Runx2 and Osterix [26]. Based on the knowledge gained from JAK and STAT knockout animals, the JAK/STAT signaling pathway was identified as important for bone development and homeostasis, recognizing that JAKs and STATs are not equally important for the biology of osteoblasts and osteoclasts. Moreover, their overall role in the musculoskeletal system is still not fully understood. Understanding the underlying mechanisms of how bone remodeling is regulated, how metabolic processes take place, and how bone responds to mechanical stimulation is central to maintaining the integrity of the skeletal system, thus ensuring human health care. Table 1 summarizes the influence of the JAK/STAT pathway in bone development using knockout animals.
Table 1. JAK/STAT pathway in bone development.
| Model System | Genes Modified | Species | Bone Phenotype | References |
|---|---|---|---|---|
| Janus kinases (JAKs) | ||||
| Jak1−/− | Jak1 deletion | Mouse | Small bone mass in contrast to wild-type mice; Perinatal lethal; Stunted embryos; Involved in bone formation |
[27][28] |
| MMTV-Cre.Jak1fl/fl | ||||
| Jak1S645P+/− | Jak1 activation | Mouse | Low bone mass levels in trabecular and cortical bone; Bone formation and resorption is increased |
[29] |
| Tofacitinib treatment | Jak1/3 inhibition | Mouse, rat | Protected against bone resorption by inflammation | [30][31][32] |
| Ruxolitinib treatment | Jak1/2 inhibition | Mouse | Protected against age-related bone resorption | [33] |
| Jak2−/− | Jak2 deletion | Mouse | Jak2-null mice die before bone formation starts; Lethality of anemia at E12.5 (erythropoiesis is absent); Involved in bone formation |
[34][35][36] |
| Jak3−/− | Jak3 deletion | Mouse | Born normally; No gross abnormality |
[37][38] |
| Tyk2−/− | Tyk2 deletion | Mouse | Viable and fertile mice; No obvious phenotype; Involved in bone formation |
[39][40] |
| Signal transducers and activators of transcription (STATs) | ||||
| Stat1−/− | Stat1 deletion | Mouse | KO mice are indistinguishable compared to wild-type mice; Higher bone mass → osteopetrotic bone phenotype; Bone exhibits excessive osteoclastogenesis; Normal epiphyseal growth plate and longitudinal bone length; Characteristics: Pro-inflammatory, antagonize proliferation |
[41][42][43] |
| Stat2−/− | Stat2 deletion | Mouse | Viable and fertile mice; No gross abnormality |
[44] |
| Stat3−/− | Stat3 deletion in all cells | Mouse | Involved in early embryonic development; Lethality at E6.5–7.5; Selective inactivation causes osteoporosis; Surface mineralization reduced; Characteristics: Pro-proliferative, anti-inflammatory |
[45][46][47][48] |
| Hyper-IgE syndrome | Stat3-DNA binding reduced in all cells | Mouse | Low bone mineral density; Recurrent fractures; Craniofacial and skeletal abnormalities |
[49][50][51] |
| SA/SA and SA/− | Reduced Stat3 phosphorylation in all cells | Mouse | Perinatal lethality: 75%; SA/SA phenotype is normal; Stat3 phosphorylation in SA/− is reduced; Reduced skeletal size |
[52] |
| Dmp1Cre.Stat3fl/fl | Stat3 deletion in osteocytes | Mouse | Low bone mass and reduced bone formation rate; Bone formation response to mechanical forced reduced |
[53] |
| Col1α1(2.3 kb) Cre; Stat3flox/flox | Stat3 deletion in osteoblasts and osteocytes | Mouse | Low trabecular bone mass and bone formation rate reduced; Normal bone length; Bone formation response to mechanical forced reduced |
[46][47][54][55] |
| Col1α1(3.6 kb) Cre; Stat3flox/flox | Stat3 deletion in chondrocytes, osteoblasts, and osteocytes | Mouse | Skeletal size is very small with low trabecular bone mass; Bone formation rate reduced and osteoclast formation increased |
[47][55] |
| Prrx1Cre; Stat3flox/flox | Stat3 deletion in chondrocytes, osteoblasts, and osteocytes | Mouse | Skeletal size reduced; Postnatal limb curvature |
[56] |
| TCre.Stat3f/f | Stat3 deletion in mesoderm-derived cells | Mouse | Shortened limbs at birth; Postnatal limb curvature |
[56] |
| Tie2(Tek)Cre.Stat3f/f | Stat3 deletion in hematopoietic and endothelial cells | Mouse | Skeletal size and bone mass are reduced; Bone formation rate reduced with increased resorption |
[57] |
| Socs3−/− | Socs3 deletion; elevated Stat3 signaling in all cells | Mouse | Embryonic lethality | [58][59] |
| VavCre.Socs3f/f | Elevated Stat3 signaling in endothelial and hematopoietic cells | Mouse | Joint inflammation; Low bone mass; Increased osteoblast and osteoclast formation |
[60] |
| Dmp1Cre.Socs3f/f | Elevated Stat3 signaling in osteocytes | Mouse | Cortical porosity increased →delayed development of cortical bone; Increased bone formation and resorption |
[61] |
| Dmp1Cre.Socs3f/f. IL6−/− |
Elevated Stat3 signaling in osteocytes; no downstream of IL-6 | Mouse | Cortical porosity increased →delayed development of cortical bone |
[61] |
| Col2Cre.Socs3f/f | Elevated Stat3 signaling in chondrocytes, osteoblasts and osteocytes | Mouse | Cortical porosity increased; Bone size reduced |
[62] |
| Stat4−/− | Stat4−/− deletion | Mouse | Viable and fertile mice; No gross abnormality |
[63] |
| Stat5a/b−/− | Double mutation | Mouse | KO mice show obviously defective bone development; Smaller Stat5a/5b (male and female) KO mice and Stat5b (male) KO mice compared to wild-type mice |
[64][65] |
| Stat5a−/− | Stat5a deletion | Mouse | Increased bone mass; Increased trabecular bone density and cortical bone formation; Prevented age-related bone loss |
[66] |
| Cathepsin K–Cre−/−Stat5fl/fl | Osteoclast-specific deletion | Mouse | Reduced bone mass | [67] |
| Stat6−/− | Stat6 deletion | Mouse | Viable and fertile mice; No gross abnormality compared to their wild-type controls |
[68][69][70] |
All members of the JAK family—Jak1, Jak2, Jak3, and Tyk2—play a pleiotropic role in physiological processes such as bone development. While Jak1, Jak2, and Tyk2 are ubiquitary, and expressed in bone cells, Jak3 is typically expressed by hematopoietic, lymphoid, and myeloid cells as mentioned above. Among Jak1 and Jak2, Jak3 and Tyk2 deficient mice show no obvious skeletal phenotype. These findings demonstrate that both Jak3 and Tyk2 are not clinically relevant for skeletal development. Most signaling cytokines depend on Jak1, and therefore it is unsurprisingly that Jak1-null mice die perinatally and weigh 40% less than the wild-type littermates, indicating that bone growth delays without Jak1 in embryos [28][71]. On the other hand, Jak2−/− embryos are anemic and die at E12.5 before bone formation starts [72]. Unfortunately, the underlying mechanisms of how Jak1 and Jak2 affect osteoblasts and osteoclasts are of clinical relevance and highlight the importance of a deep understanding. Similar to Jaks, Stat proteins are located in bone tissue. The STAT family, first discovered in 1993 by James Darnell [73], consists of seven signal transducer and activator of transcription proteins. While Stat2, Stat4, and Stat6 do not play a crucial role in skeletal development, indicated by a normal skeletal phenotype, Stat1 is a critical regulator of both osteoclastogenesis and osteoblast differentiation. Therefore, Stat1 depletion leads to excessive osteoclastogenesis and inhibition of the transcription factor Runx2 as well as suppression of Osterix transcription in osteoblasts [43]. Although Stat1−/− mice are indistinguishable from their normal controls, depletion leads to an osteopetrotic bone phenotype characterized by an increased bone mass [42]. These findings suggest that Stat1 has negative effects on bone formation in vivo. Based on the normal epiphyseal growth plate, Kim et al. suggest that physiological chondrocyte proliferation is not significantly increased due to Stat1 depletion [42]. Among the seven, Stat3, Stat5a, and Stat5b have been shown to be directly involved in bone development. Stat3 was first described as a DNA-binding protein that is activated in IL-6-stimulated hepatocytes [74]. In humans, STAT3 is probably the most important transcription factor. Studies suggest that Stat3 plays a central role in early embryonic bone formation, is involved in bone metabolism, and reduces mechanical load-driven bone development [46][47]. Since Stat3 mediates intracellular signal transduction in osteoblasts and osteoclasts, depletion reduces bone mass and impairs bone development. Thus, the incidence of bone fractures increases [46][47]. Along with other members of the STAT family, Stat5 was originally identified as a cytosolic signal molecule involved in the proliferation, differentiation, and progression of solid tumor cells [75]. Recent evidence suggests that STATs, especially Stat5 play a central role in growth hormone signaling, osteoblast differentiation, inhibition of osteoclast differentiation, and therefore bone homeostasis [76][77]. The depletion of both Stat5a and Stat5b in mice therefore lead to apparently defective bone formation in vivo. This delayed skeletal development is consistent with insulin like growth factor (IGF)-1 function in bone, which were significantly reduced by Stat5a/b mutation [67]. Moreover, the genetic mapping of the STAT gene family should be comment. Indeed, studies suggest that Stat1, Stat2, Stat3, Stat4, and Stat6 arose by chromosome duplications from Stat5 [78]. Therefore, both Stat5a and Stat5b show extensive similarities regarding their sequence with isoform-specific functions. Deletion of Stat5a leads to increased bone mineral density, trabecular and cortical bone mass and prevents age-related bone loss in mice [66]. Lee et al. investigated the role of STAT5a in human bone marrow-derived mesenchymal stromal cells. Surprisingly, inhibition of STAT5a resulted in a significant increase of osteoblast differentiation, whereas inhibition of STAT5b showed no effect. This demonstrates the isoform-specific function of the STAT5s. In addition, STAT5b has been shown to apparently regulate the male pattern of long bone growth that is characteristic of many species, including humans [65]. Nevertheless, further studies are needed to gain a better understanding on the detailed mode of action.
References
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-Stat Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; O’Shea, J.J. Jaks and Stats: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling Via the Jak/Stat Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.L.; Holland, S.M.; Notarangelo, L.D. Inborn Errors of Human Jaks and Stats. Immunity 2012, 36, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A. The Jak1/Stat3/Socs3 Axis in Bone Development, Physiology, and Pathology. Exp. Mol. Med. 2020, 52, 1185–1197. [Google Scholar] [CrossRef]
- Onan, D.; Allan, E.H.; Quinn, J.M.; Gooi, J.H.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Martin, T.J. The Chemokine Cxcl1 Is a Novel Target Gene of Parathyroid Hormone (Pth)/Pth-Related Protein in Committed Osteoblasts. Endocrinology 2009, 150, 2244–2253. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Gubrij, I.; Lin, S.C.; Saylors, R.L.; Manolagas, S.C. Stat3 Activation in Stromal/Osteoblastic Cells Is Required for Induction of the Receptor Activator of Nf-Kappab Ligand and Stimulation of Osteoclastogenesis by Gp130-Utilizing Cytokines or Interleukin-1 but Not 1,25-Dihydroxyvitamin D3 or Parathyroid Hormone. J. Biol. Chem. 1999, 274, 19301–19308. [Google Scholar]
- Sims, N.A. Cell-Specific Paracrine Actions of Il-6 Family Cytokines from Bone, Marrow and Muscle That Control Bone Formation and Resorption. Int. J. Biochem. Cell Biol. 2016, 79, 14–23. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M Promotes Bone Formation Independently of Resorption When Signaling through Leukemia Inhibitory Factor Receptor in Mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef]
- Ni, L.; Yu, J.; Gui, X.; Lu, Z.; Wang, X.; Guo, H.; Zhou, Y. Overexpression of Rpn2 Promotes Osteogenic Differentiation of Hbmscs through the Jak/Stat3 Pathway. FEBS Open Bio 2020, 10, 158–167. [Google Scholar] [CrossRef]
- McGregor, N.E.; Murat, M.; Elango, J.; Poulton, I.J.; Walker, E.C.; Crimeen-Irwin, B.; Ho, P.W.M.; Gooi, J.H.; Martin, T.J.; Sims, N.A. Il-6 Exhibits Both Cis- and Trans-Signaling in Osteocytes and Osteoblasts, but Only Trans-Signaling Promotes Bone Formation and Osteoclastogenesis. J. Biol. Chem. 2019, 294, 7850–7863. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Brennan, H.J.; Vrahnas, C.; Poulton, I.J.; McGregor, N.E.; Standal, T.; Walker, E.C.; Koh, T.T.; Nguyen, H.; Walsh, N.C.; et al. The Primary Function of Gp130 Signaling in Osteoblasts Is to Maintain Bone Formation and Strength, Rather Than Promote Osteoclast Formation. J. Bone Miner. Res. 2014, 29, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Poulton, I.J.; McGregor, N.E.; Pompolo, S.; Walker, E.C.; Sims, N.A. Contrasting Roles of Leukemia Inhibitory Factor in Murine Bone Development and Remodeling Involve Region-Specific Changes in Vascularization. J. Bone Miner. Res. 2012, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 Is an Osteoclast-Derived Stimulus of Bone Formation Required for Normal Bone Remodeling. J. Bone Miner. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 Inhibitor, Baricitinib, Inhibits Osteoclastogenesis by Suppressing Rankl Expression in Osteoblasts in Vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [PubMed]
- Fujio, Y.; Maeda, M.; Mohri, T.; Obana, M.; Iwakura, T.; Hayama, A.; Yamashita, T.; Nakayama, H.; Azuma, J. Glycoprotein 130 Cytokine Signal as a Therapeutic Target against Cardiovascular Diseases. J. Pharmacol. Sci. 2011, 117, 213–222. [Google Scholar] [CrossRef]
- Nur, H.; Rao, L.; Frassanito, M.A.; De Raeve, H.; Ribatti, D.; Mfopou, J.K.; Van Valckenborgh, E.; De Bruyne, E.; Vacca, A.; Vanderkerken, K.; et al. Stimulation of Invariant Natural Killer T Cells by Alpha-Galactosylceramide Activates the Jak-Stat Pathway in Endothelial Cells and Reduces Angiogenesis in the 5t33 Multiple Myeloma Model. Br. J. Haematol. 2014, 167, 651–663. [Google Scholar] [CrossRef]
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a Patient with Scid: Essential Role of Jak3 in Lymphoid Development. Science 1995, 270, 797–800. [Google Scholar] [CrossRef]
- Macchi, P.; Villa, A.; Giliani, S.; Sacco, M.G.; Frattini, A.; Porta, F.; Ugazio, A.G.; Johnston, J.A.; Candotti, F.; O’Shea, J.J.; et al. Mutations of Jak-3 Gene in Patients with Autosomal Severe Combined Immune Deficiency (Scid). Nature 1995, 377, 65–68. [Google Scholar] [CrossRef]
- Zhang, D.; Wlodawer, A.; Lubkowski, J. Crystal Structure of a Complex of the Intracellular Domain of Interferon Lambda Receptor 1 (Ifnlr1) and the Ferm/Sh2 Domains of Human Jak1. J. Mol. Biol. 2016, 428, 4651–4668. [Google Scholar] [CrossRef]
- Floss, D.M.; Klocker, T.; Schroder, J.; Lamertz, L.; Mrotzek, S.; Strobl, B.; Hermanns, H.; Scheller, J. Defining the Functional Binding Sites of Interleukin 12 Receptor Beta1 and Interleukin 23 Receptor to Janus Kinases. Mol. Biol. Cell 2016, 27, 2301–2316. [Google Scholar] [CrossRef] [PubMed]
- Wallweber, H.J.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural Basis of Recognition of Interferon-Alpha Receptor by Tyrosine Kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Skiniotis, G.; Rice, A.J.; Thomas, C.; Fischer, S.; Walz, T.; Garcia, K.C. Structural Snapshots of Full-Length Jak1, a Transmembrane Gp130/Il-6/Il-6ralpha Cytokine Receptor Complex, and the Receptor-Jak1 Holocomplex. Structure 2011, 19, 45–55. [Google Scholar] [CrossRef]
- Kido, S.; Kuriwaka-Kido, R.; Imamura, T.; Ito, Y.; Inoue, D.; Matsumoto, T. Mechanical Stress Induces Interleukin-11 Expression to Stimulate Osteoblast Differentiation. Bone 2009, 45, 1125–1132. [Google Scholar] [CrossRef]
- Ahlen, J.; Andersson, S.; Mukohyama, H.; Roth, C.; Backman, A.; Conaway, H.H.; Lerner, U.H. Characterization of the Bone-Resorptive Effect of Interleukin-11 in Cultured Mouse Calvarial Bones. Bone 2002, 31, 242–251. [Google Scholar] [CrossRef]
- Zhu, J.; Shimizu, E.; Zhang, X.; Partridge, N.C.; Qin, L. Egfr Signaling Suppresses Osteoblast Differentiation and Inhibits Expression of Master Osteoblastic Transcription Factors Runx2 and Osterix. J. Cell Biochem. 2011, 112, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Sakamoto, K.; Wehde, B.L.; Radler, P.D.; Triplett, A.A.; Wagner, K.U. Generation of Janus Kinase 1 (Jak1) Conditional Knockout Mice. Genesis 2016, 54, 582–588. [Google Scholar] [CrossRef]
- Sabrautzki, S.; Janas, E.; Lorenz-Depiereux, B.; Calzada-Wack, J.; Aguilar-Pimentel, J.A.; Rathkolb, B.; Adler, T.; Cohrs, C.; Hans, W.; Diener, S.; et al. An Enu Mutagenesis-Derived Mouse Model with a Dominant Jak1 Mutation Resembling Phenotypes of Systemic Autoimmune Disease. Am. J. Pathol. 2013, 183, 352–368. [Google Scholar] [CrossRef]
- Mori, T.; Miyamoto, T.; Yoshida, H.; Asakawa, M.; Kawasumi, M.; Kobayashi, T.; Morioka, H.; Chiba, K.; Toyama, Y.; Yoshimura, A. Il-1beta and Tnfalpha-Initiated Il-6-Stat3 Pathway Is Critical in Mediating Inflammatory Cytokines and Rankl Expression in Inflammatory Arthritis. Int. Immunol. 2011, 23, 701–712. [Google Scholar] [CrossRef]
- LaBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. Jak Inhibition with Tofacitinib Suppresses Arthritic Joint Structural Damage through Decreased Rankl Production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Cascao, R.; Finnila, M.A.J.; Lopes, I.P.; da Gloria, V.G.; Saarakkala, S.; Zioupos, P.; Canhao, H.; Fonseca, J.E. Effects of Tofacitinib in Early Arthritis-Induced Bone Loss in an Adjuvant-Induced Arthritis Rat Model. Rheumatology 2019, 58, 371. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef]
- Gotthardt, D.; Trifinopoulos, J.; Sexl, V.; Putz, E.M. Jak/Stat Cytokine Signaling at the Crossroad of Nk Cell Development and Maturation. Front. Immunol. 2019, 10, 2590. [Google Scholar] [CrossRef]
- Krempler, A.; Qi, Y.; Triplett, A.A.; Zhu, J.; Rui, H.; Wagner, K.U. Generation of a Conditional Knockout Allele for the Janus Kinase 2 (Jak2) Gene in Mice. Genesis 2004, 40, 52–57. [Google Scholar] [CrossRef]
- Park, S.Y.; Saijo, K.; Takahashi, T.; Osawa, M.; Arase, H.; Hirayama, N.; Miyake, K.; Nakauchi, H.; Shirasawa, T.; Saito, T. Developmental Defects of Lymphoid Cells in Jak3 Kinase-Deficient Mice. Immunity 1995, 3, 771–782. [Google Scholar] [CrossRef]
- Nosaka, T.; van Deursen, J.M.; Tripp, R.A.; Thierfelder, W.E.; Witthuhn, B.A.; McMickle, A.P.; Doherty, P.C.; Grosveld, G.C.; Ihle, J.N. Defective Lymphoid Development in Mice Lacking Jak3. Science 1995, 270, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Kuo, F.I.; Smith, P.A. Targeting the Janus Kinase Family in Autoimmune Skin Diseases. Front. Immunol. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the Jak-Stat Signaling Pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Kim, S.; Koga, T.; Isobe, M.; Kern, B.E.; Yokochi, T.; Chin, Y.E.; Karsenty, G.; Taniguchi, T.; Takayanagi, H. Stat1 Functions as a Cytoplasmic Attenuator of Runx2 in the Transcriptional Program of Osteoblast Differentiation. Genes Dev. 2003, 17, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Takaishi, H.; Takito, J.; Tohmonda, T.; Yoda, M.; Ota, N.; Kosaki, N.; Matsumoto, M.; Ikegami, H.; Nakamura, T.; et al. Inhibition of Stat1 Accelerates Bone Fracture Healing. J. Orthop. Res. 2010, 28, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune Response in Stat2 Knockout Mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted Disruption of the Mouse Stat3 Gene Leads to Early Embryonic Lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Itoh, S.; Udagawa, N.; Takahashi, N.; Yoshitake, F.; Narita, H.; Ebisu, S.; Ishihara, K. A Critical Role for Interleukin-6 Family-Mediated Stat3 Activation in Osteoblast Differentiation and Bone Formation. Bone 2006, 39, 505–512. [Google Scholar] [CrossRef]
- Zhou, H.; Newnum, A.B.; Martin, J.R.; Li, P.; Nelson, M.T.; Moh, A.; Fu, X.Y.; Yokota, H.; Li, J. Osteoblast/Osteocyte-Specific Inactivation of Stat3 Decreases Load-Driven Bone Formation and Accumulates Reactive Oxygen Species. Bone 2011, 49, 404–411. [Google Scholar] [CrossRef]
- Atsumi, T.; Ishihara, K.; Kamimura, D.; Ikushima, H.; Ohtani, T.; Hirota, S.; Kobayashi, H.; Park, S.J.; Saeki, Y.; Kitamura, Y.; et al. A Point Mutation of Tyr-759 in Interleukin 6 Family Cytokine Receptor Subunit Gp130 Causes Autoimmune Arthritis. J. Exp. Med. 2002, 196, 979–990. [Google Scholar] [CrossRef]
- Yong, P.F.; Freeman, A.F.; Engelhardt, K.R.; Holland, S.; Puck, J.M.; Grimbacher, B. An Update on the Hyper-Ige Syndromes. Arthritis Res. Ther. 2012, 14, 228. [Google Scholar] [CrossRef]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. Stat3 Mutations in the Hyper-Ige Syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-Negative Mutations in the DNA-Binding Domain of Stat3 Cause Hyper-Ige Syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential Role of Stat3 in Postnatal Survival and Growth Revealed by Mice Lacking Stat3 Serine 727 Phosphorylation. Mol. Cell Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Corry, K.A.; Zhou, H.; Brustovetsky, T.; Himes, E.R.; Bivi, N.; Horn, M.R.; Kitase, Y.; Wallace, J.M.; Bellido, T.; Brustovetsky, N.; et al. Stat3 in Osteocytes Mediates Osteogenic Response to Loading. Bone Rep. 2019, 11, 100218. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Sun, Y.L.; Hoey, T.; Grusby, M.J. Impaired Il-12 Responses and Enhanced Development of Th2 Cells in Stat4-Deficient Mice. Nature 1996, 382, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Woitge, H.W.; Braut, A.; Kronenberg, M.S.; Lichtler, A.C.; Mina, M.; Kream, B.E. Expression and Activity of Osteoblast-Targeted Cre Recombinase Transgenes in Murine Skeletal Tissues. Int. J. Dev. Biol. 2004, 48, 645–653. [Google Scholar] [CrossRef]
- Hall, M.D.; Murray, C.A.; Valdez, M.J.; Perantoni, A.O. Mesoderm-Specific Stat3 Deletion Affects Expression of Sox9 Yielding Sox9-Dependent Phenotypes. PLoS Genet. 2017, 13, e1006610. [Google Scholar] [CrossRef]
- Zhang, Z.; Welte, T.; Troiano, N.; Maher, S.E.; Fu, X.Y.; Bothwell, A.L. Osteoporosis with Increased Osteoclastogenesis in Hematopoietic Cell-Specific Stat3-Deficient Mice. Biochem. Biophys. Res. Commun. 2005, 328, 800–807. [Google Scholar] [CrossRef]
- Marine, J.C.; McKay, C.; Wang, D.; Topham, D.J.; Parganas, E.; Nakajima, H.; Pendeville, H.; Yasukawa, H.; Sasaki, A.; Yoshimura, A.; et al. Socs3 Is Essential in the Regulation of Fetal Liver Erythropoiesis. Cell 1999, 98, 617–627. [Google Scholar] [CrossRef]
- Roberts, A.W.; Robb, L.; Rakar, S.; Hartley, L.; Cluse, L.; Nicola, N.A.; Metcalf, D.; Hilton, D.J.; Alexander, W.S. Placental Defects and Embryonic Lethality in Mice Lacking Suppressor of Cytokine Signaling 3. Proc. Natl. Acad. Sci. USA 2001, 98, 9324–9329. [Google Scholar] [CrossRef]
- Wong, P.K.; Egan, P.J.; Croker, B.A.; O’Donnell, K.; Sims, N.A.; Drake, S.; Kiu, H.; McManus, E.J.; Alexander, W.S.; Roberts, A.W.; et al. Socs-3 Negatively Regulates Innate and Adaptive Immune Mechanisms in Acute Il-1-Dependent Inflammatory Arthritis. J. Clin. Investig. 2006, 116, 1571–1581. [Google Scholar] [CrossRef]
- Cho, D.C.; Brennan, H.J.; Johnson, R.W.; Poulton, I.J.; Gooi, J.H.; Tonkin, B.A.; McGregor, N.E.; Walker, E.C.; Handelsman, D.J.; Martin, T.J.; et al. Bone Corticalization Requires Local Socs3 Activity and Is Promoted by Androgen Action Via Interleukin-6. Nat. Commun. 2017, 8, 806. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; D’Cruz, A.A.; Hansen, J.; Croker, B.A.; Lawlor, K.E.; Sims, N.A.; Wicks, I.P. Deleting Suppressor of Cytokine Signaling-3 in Chondrocytes Reduces Bone Growth by Disrupting Mitogen-Activated Protein Kinase Signaling. Osteoarthr. Cartil. 2019, 27, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, W.E.; van Deursen, J.M.; Yamamoto, K.; Tripp, R.A.; Sarawar, S.R.; Carson, R.T.; Sangster, M.Y.; Vignali, D.A.; Doherty, P.C.; Grosveld, G.C.; et al. Requirement for Stat4 in Interleukin-12-Mediated Responses of Natural Killer and T Cells. Nature 1996, 382, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a Is Mandatory for Adult Mammary Gland Development and Lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of Stat5b for Sexual Dimorphism of Body Growth Rates and Liver Gene Expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Park, K.H.; Hwang, J.S.; Lee, M.; Yoon, D.S.; Ryu, H.A.; Jung, H.S.; Park, K.W.; Kim, J.; Park, S.W.; et al. Inhibition of Stat5a Promotes Osteogenesis by Dlx5 Regulation. Cell Death Dis 2018, 9, 1136. [Google Scholar] [CrossRef] [PubMed]
- Hirose, J.; Masuda, H.; Tokuyama, N.; Omata, Y.; Matsumoto, T.; Yasui, T.; Kadono, Y.; Hennighausen, L.; Tanaka, S. Bone Resorption Is Regulated by Cell-Autonomous Negative Feedback Loop of Stat5-Dusp Axis in the Osteoclast. J. Exp. Med. 2014, 211, 153–163. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 Is Required for Mediating Responses to Il-4 and for Development of Th2 Cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential Role of Stat6 in Il-4 Signalling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef]
- Shimoda, K.; van Deursen, J.; Sangster, M.Y.; Sarawar, S.R.; Carson, R.T.; Tripp, R.A.; Chu, C.; Quelle, F.W.; Nosaka, T.; Vignali, D.A.; et al. Lack of Il-4-Induced Th2 Response and Ige Class Switching in Mice with Disrupted Stat6 Gene. Nature 1996, 380, 630–633. [Google Scholar] [CrossRef]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of Fda-Approved Anti-Inflammatory Drugs against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, H.; Cumano, A.; Muller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 Deficiency Defines an Essential Developmental Checkpoint in Definitive Hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E., Jr. A Single Phosphotyrosine Residue of Stat91 Required for Gene Activation by Interferon-Gamma. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef] [PubMed]
- Raz, R.; Durbin, J.E.; Levy, D.E. Acute Phase Response Factor and Additional Members of the Interferon-Stimulated Gene Factor 3 Family Integrate Diverse Signals from Cytokines, Interferons, and Growth Factors. J. Biol. Chem. 1994, 269, 24391–24395. [Google Scholar]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/B with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef]
- Lee, J.; Seong, S.; Kim, J.H.; Kim, K.; Kim, I.; Jeong, B.C.; Nam, K.I.; Kim, K.K.; Hennighausen, L.; Kim, N. Stat5 Is a Key Transcription Factor for Il-3-Mediated Inhibition of Rankl-Induced Osteoclastogenesis. Sci. Rep. 2016, 6, 30977. [Google Scholar] [CrossRef]
- Darvin, P.; Joung, Y.H.; Yang, Y.M. Jak2-Stat5b Pathway and Osteoblast Differentiation. JAKSTAT 2013, 2, e24931. [Google Scholar] [CrossRef]
- Wang, Y.; Levy, D.E. Comparative Evolutionary Genomics of the Stat Family of Transcription Factors. JAKSTAT 2012, 1, 23–33. [Google Scholar] [CrossRef]

