 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Niarakis | + 2198 word(s) | 2198 | 2021-01-07 09:31:21 | | | |
| 2 | Lily Guo | Meta information modification | 2198 | 2021-01-18 12:28:13 | | |
Video Upload Options
Fibroblasts, the most abundant cells in the connective tissue, are key modulators of the extracellular matrix (ECM) composition.
1. Introduction
Fibroblasts were initially described during the 19th century by Virchow [1] and Duval[2] as the most common cell type from connective tissue. They also exhibit a round, large pale and flat nucleus with prominent nucleoli, indicating a very active RNA metabolism[3]. Fibroblasts are known to be essential for a significant number of physiological functions. They produce extracellular matrix (ECM) proteins (e.g., collagen, glycosaminoglycans, fibronectin, laminins, and proteoglycans) and produce the structural framework—stroma—for tissues[4]. They induce epithelial differentiation, regulate inflammation[5], and play a critical role in wound healing by migrating to the damaged tissue [6]. Fibroblasts are widely known to display remarkable phenotypic plasticity with the ability to adapt quickly and efficiently to their environment when activated by appropriate stimuli. For instance, it has been acknowledged that fibroblasts can play a significant role in disease pathogenesis by presenting complex phenotypes and functions according to the biological context. Indeed, some fibroblasts (e.g., gingival[7], dermal [8], lung [9], cardiac[10] and synovial fibroblasts [11]) can express innate immune receptors to sense pathogens and present antigens, contributing to the immune response [12].
A certain type of fibroblasts, secreting myofibroblasts, play a central role in fibrosis. Fibrosis is the common endpoint of many chronic inflammatory diseases and includes the excessive deposit of fibrous connective tissue and ECM molecules such as collagen and fibronectin, in and around damaged tissue[13]. Besides inflammatory diseases, fibrosis is a pathological trait of chronic autoimmune diseases, such as Rheumatoid Arthritis (RA), Crohn’s disease, myelofibrosis and systemic lupus erythematosus to name a few, and can also affect tumor invasion and metastasis in cancer conditions[14].
Nevertheless, relatively few studies have considered regulating fibroblasts’ functions, either in inflammatory or autoimmune diseases, by developing new therapeutic targets. More efforts are needed to understand the critical role of fibroblasts in disease pathogenesis, focusing on the shared characteristics that seemingly drive disease onset and progression in a variety of pathological conditions[15].
2. The Role of Fibroblasts in Rheumatoid Arthritis
In the joint synovium, fibroblasts represent the primary stromal cells. They ensure the structural integrity of synovial sub-lining and lining by forming a layer thick as one or two cells, interspersed with tissue-resident macrophages [16]. Fibroblasts guarantee nutrient supply and secrete hyaluronic acid and lubricin (two essential constituents of synovial fluid) responsible for lubricating the joints[17][18]. They are also responsible for producing the nonrigid ECM of the synovial fluid, rich in type 1 and type 2 collagen, helping wound healing and damaged tissue reparation[12]. Many studies in RA focus on fibroblasts, as these cells play a significant role in disease pathogenesis.
RA is an autoimmune disease with a prevalence of approximately 0.5% to 1% in the population. The onset of the disease is characterized by the pannus formation, consisting of the hyperplastic synovium due to the lay down of synovial macrophages and fibroblasts (RASFs). The pannus is highly invasive and has destructive effects on the adjacent cartilage tissue and bone[4][19][20][21]. Synovial fibroblasts in RA exhibit different characteristics from healthy fibroblasts in terms of morphology and gene expression. The stressful environment created in the inflamed joint in combination with nutrient competition leads fibroblasts to adopt a more aggressive phenotype to ensure survival (Figure 1).
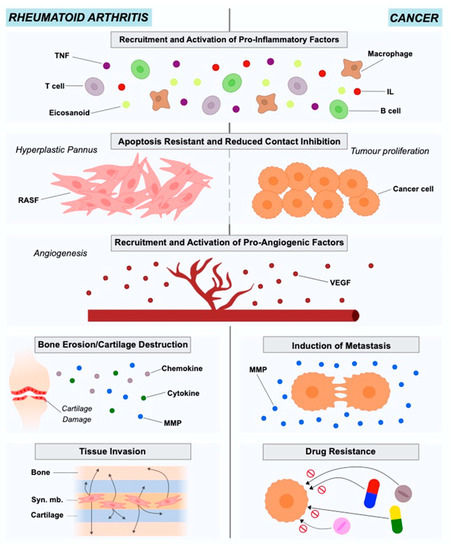
Figure 1. Roles of rheumatoid arthritis synovial fibroblasts and cancer-associated fibroblasts in in rheumatoid arthritis and cancer pathogenesis and progression.
At this point, RASFs have reduced contact inhibition, express altered levels of adhesion molecules, cytokines, chemokines and matrix-degrading enzymes, causing cartilage damage and mediating the interaction with neighboring inflammatory and endothelial cells, affecting the bone via regulation of monocyte to osteoclast differentiation[22]. RASFs support the development of the hyperplastic RA synovium as tertiary lymphoid organs (TLOs) by interacting with immune cells like T cells and B cells, producing several mediators and organizing ectopic (tertiary) lymphoid-like structures (ELSs)[23]. They are also resistant to apoptosis and have an increased ability to migrate and invade periarticular tissues, including bone and cartilage, contributing to their destruction[11][21]. RASFs can also be considered to be primary drivers of inflammation, angiogenesis and cell growth[21]. They disturb the homeostatic balance between leukocyte recruitment, proliferation, emigration and death, leading to a persistent leukocyte infiltration[22]. In this way, RASFs are no longer considered to be passive bystanders, but as active players in RA pathogenesis and sustained chronicity, and RASF-directed therapies could become a complementary approach to currently used immune-focused therapies.
2.1. Origin of Rheumatoid Arthritis Synovial Fibroblasts
The origin of RASFs remains elusive. In earlier studies, researchers found that CD34(+) cells in RA patients are regulated by TNF∝
and can differentiate into fibroblast-like cells, suggesting that bone marrow CD34+ could be the origin of RASFs[24]. Presently, however, it has been suggested that RASFs descent from mesenchymal stem cells and possess some typical fibroblast markers, such as ICAM1 integrins, the surface marker Thy-1 (CD90), and type IV and V collagens. In recent studies, researchers used lineage-tracing of Gdf5+ mesenchymal stromal/stem cells in the synovial tissue and their findings supported this hypothesis regarding the ontogeny of the RASFs[25]. Regarding markers that can be found in fibroblasts, vimentin and α-smooth muscle actin seem to be more generic while UDP-glucose 6-dehydrogenase, vascular cell adhesion molecule-1, and cadherin-11 (CDH11) are found to be fibroblast specific[26][27].
2.2. Population Heterogeneity in Rheumatoid Arthritis Synovial Fibroblasts
Recent studies benefiting from the advancements in single-cell RNA sequencing (scRNA-Seq) technologies and bioinformatics methodologies, have documented the presence of distinct subsets of fibroblasts’ subpopulations in arthritis which are responsible for mediating distinct pathological traits such as inflammation and tissue damage.
Mizoguchi et al. [28], studied the functional and transcriptional differences between fibroblast subsets in human synovial tissues from RA and osteoarthritis (OA) patients using bulk and single-cell transcriptomics. They succeeded in identifying seven fibroblast subsets with distinct surface protein phenotypes. These seven subpopulations were subsequently collapsed into three subsets by integrating transcriptomic data. The findings of this study showed that a distinct fibroblast subset expressing podoplanin, THY1 membrane glycoprotein and cadherin-11, but lacking CD34, is three times more elevated in RA patients in comparison to OA patients. This subset that is anatomically located in the perivascular zone of the synovium, can secrete proinflammatory cytokines, has a high proliferation rate, and present an in vitro invasive phenotype. Croft et al. [29], used mouse models of persistent arthritis to study the deletion of the fibroblast activation protein-α (FAPα) in fibroblasts and showed that such a deletion suppressed both inflammation and bone erosions. The use of single-cell transcriptional analysis allowed the identification of two distinct fibroblast subsets: the FAPα+THY1+ subpopulation that is located in the synovial sub-lining and plays the role of the immune effector, and the FAPα+THY1− subpopulation positioned in the lining layer of the synovium which exhibits destructive properties. When transferred into the joint, the first subpopulation, would boost inflammation, whereas the second regulated predominantly bone and cartilage damage. Recently, in the meta-analysis study of Zerrouk et al.[30], researchers estimated the different transcriptional factor activities between RA and OA fibroblasts using gene expression data and network inference. In this study, the transcriptional factor profiles of the seven subpopulations of the Mizoguchi study[28] were calculated and compared to the corresponding OA subpopulations highlighting differences between RA and OA, but also among the subsets if the same pathological condition.
2.3. Epigenetic Modifications in Rheumatoid Arthritis Synovial Fibroblasts
RASFs epigenetic profile can help to understand RA pathogenesis better and to identify new therapeutic targets[31]. Karouzakis et al. [32] showed that RASFs have a hypomethylated genome, with several hypomethylated genes playing a role in their main characteristics, such as extracellular matrix interactions, adhesion and cell migration. In a more recent study results showed that the expression of DNA (cytosine-5)-methyltransferase 1 (DNMT1) is lower in RASFs as compared with OA FLSs while the components of polyamine metabolism were higher[31]. While histone methylation mechanisms are not well understood, histone acetylation is better characterized in RASFs. HDAC3 (histone deacetylase 3) is a potential key player for inflammation inhibition in RA disease. It has been shown that HDAC3 suppresses inflammation in RASFs as much as pan-HDAC inhibition, and there seems to be a significant difference in histone acetylation in RASFs as compared to OA fibroblast-like synoviocytes[31].
3. The Role of Fibroblasts in Cancer
The tumor microenvironment (TME), known also as the tumor stroma, comprises the ECM, blood vessels, endothelial and stromal cells (e.g., fibroblasts), and also immune cells[33][34]. The importance of TME in cancer onset and progression has been highlighted for years. Cancer is the result of genetic and epigenetic alterations in clonal cells. The regulation of these altered clonal cells regarding survival, growth and metastasis is under the control of the interactions between cancer and TME cells [35]. Studies in different cancer types such as lung, prostate, breast and colon have indicated Cancer-Associated Fibroblasts (CAFs) as active players in disease initiation and also as important contributors to tumor growth, survival and invasion[36] (Figure 1).
Several studies using in vitro experimentation have provided evidence on the role of CAFs in cancer progression. Studies using mouse models suggest that CAFs are capable of promoting cell proliferation, angiogenesis, tissue invasion and metastasis. More specifically, the tumors that are formed after transplantation of cancer cells and CAFs are more malignant than the tumors formed when transplantation involved cancer cells alone or cancer cells with healthy fibroblasts[37][38]. Lastly, co-implantation of CAFs along with pro-malignant prostate cancer cells resulted in the malignant transition and proliferation of the prostate cells[39].
The hypotheses proposed to explain the transition of the healthy fibroblasts to CAFs involve autocrine and paracrine mechanisms for the secretion of cytokines, chemokines, and growth factors by the stromal cells. These mediators will in turn regulate gene expression through specific signaling cascades and will contribute to the expression of a metastatic cancer type with elevated growth rates and invasiveness[36][40][41][42][43][44][45].
3.1. Origin of Cancer-Associated Fibroblasts
The precise origin of CAFs is a debated subject. Several potential sources have been proposed throughout the years such as healthy fibroblasts, epithelial or endothelial cells, Mesenchymal Stem Cells (MSCs), as well as Smooth Muscle Cells (SMCs)[46] [46]. Indeed, the obvious hypothesis lies in an alteration of local precursors (i.e., healthy fibroblasts) following too much exposure to cancer cells, transforming them into CAFs[46][47]. Nevertheless, the TME being composed of both epithelial and endothelial cells, they are also considered to be a potential source of CAFs. Epithelial cells are known to show plasticity and epithelial-to-mesenchymal (EMT) transition is suspected to be at the origin of CAFs[48]. In addition, Zeisberg et al.[49] support the hypothesis that endothelial cells, treated specifically, can demonstrate CAF morphology and phenotype. Several reports also support the assumption that MSCs, apart from aggravating tumors proliferation, invasion and metastasis [50], are a potential origin for CAFs: Quante’s use of murine models revealed that at least 20% of CAFs derived from MSCs [51], whereas Direkze et al. approximated this proportion to 25% [52]. Finally, Wikstrom et al.[53] believe that differentiated SMCs can be at the origin of CAFs in prostate tumors.
3.2. Population Heterogeneity in Cancer-Associated Fibroblasts
Understanding the heterogeneity of the cells belonging to the TME is essential for elucidating complex mechanisms and designing novel strategies for precision medicine. CAFs present a heterogeneous population and a detailed study and classification of the roles, functions, traits of the CAFs subsets is critical for designing CAF-targeted therapies[54]. ScRNA-Seq technologies could help shed light onto the population heterogeneity of cancer and cancer-associated cells for a wide range of cancer types.
To date, many studies focusing on the heterogeneity of CAFs in various cancer types have been published. We will focus on breast cancer CAF heterogeneity studies to provide an example, but the reader can find more information on other cancer types in dedicated reviews[54].
Bartoschek et al.[55], using scRNA-Seq data of 768 mesenchymal cells transcriptomes from a breast cancer mouse model, defined three distinct CAF subpopulations that could be attributed to distinct anatomical positions. Moreover, gene profiles of CAF subtypes were shown to correlate with characteristic functional programs, suggesting that biomarker signatures of each subpopulation could be achievable. Sebastian et al. [56], studied the molecular and phenotypic heterogeneity of CAFs in triple-negative breast cancer (TNBC) using a syngeneic mouse model, BALB/c-derived 4T1 mammary tumors. Using scRNA-Seq they were able to identify six CAF subpopulations in 4T1 tumors, with three subpopulations also present in CAFs from pancreatic cancer. Their study also showed that some of the cells identified were present in normal breast/pancreas tissue, revealing phenotypes that are not TME-induced. In a complementary study on breast cancer by Kieffer et al. [57], researchers identified 8 CAF-S1 clusters by analyzing more than 19,000 single CAF-S1 fibroblasts from breast cancer. Using flow cytometry and in-silico analyses their study highlights a positive feedback loop between specific CAF-S1 clusters and Tregs and uncovers their role in immunotherapy resistance.
3.3. Epigenetic Alterations of Cancer-Associated Fibroblasts
CAFs do not acquire somatic mutations, therefore other mechanisms, such as epigenetic regulation are being investigated in several types of cancer as potentially responsible for their phenotypic transformation, development, and acquisition of tumor supportive features. Such epigenetic regulations involve post-transcriptional control by miRNAs acting as oncogene and/or tumor suppressor through various target genes[58] in breast cancer [59] and bladder cancer [60], DNA methylation to activate and overexpress oncogenes[58] in prostate cancer[61] as well as colorectal cancer [62], and finally chromatin and histone modification to inhibit or down-regulate tumor suppressor genes[63] in prostate cancer for example[64].
References
- Virchow: R. Die Cellularpathologie in ihrer Begrundung auf Physiologische und Pathologische Gewebelehre; Hirschwald, A.: Berlin, Germany, 1862.
- Duval, M. Atlas D’embryologie; Masson G.: Paris, France, 1889.
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675, doi:10.1093/rheumatology/kel065.
- Wegner, N.; Lundberg, K.; Kinloch, A.; Fisher, B.; Malmström, V.; Feldmann, M.; Venables, P.J. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010, 233, 34–54, doi:10.1111/j.0105-2896.2009.00850.x.
- Croft, C.B.; Tarin, D. Ultrastructural studies of wound healing in mouse skin. I. Epithelial behaviour. J. Anat. 1970, 106, 63–77.
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–412, doi:10.12968/jowc.2013.22.8.407.
- Pinheiro, C.R.; Coelho, A.L.; de Oliveira, C.E.; Gasparoto, T.H.; Garlet, G.P.; Silva, J.S.; Santos, C.F.; Cavassani, K.A.; Ho-gaboam, C.M.; Campanelli, A.P. Recognition of Candida albicans by gingival fibroblasts: The role of TLR2, TLR4/CD14, and MyD88. Cytokine 2018, 106, 67–75, doi:10.1016/j.cyto.2017.10.013.
- Bellei, B.; Caputo, S.; Carbone, A.; Silipo, V.; Papaccio, F.; Picardo, M.; Eibenschutz, L. The Role of Dermal Fibroblasts in Nevoid Basal Cell Carcinoma Syndrome Patients: An Overview. Int. J. Mol. Sci. 2020, 21, 720, doi:10.3390/ijms21030720.
- Para, R.; Romero, F.; George, G.; Summer, R. Metabolic Reprogramming as a Driver of Fibroblast Activation in Pulmo-naryFibrosis. Am. J. Med. Sci. 2019, 357, 394–398, doi:10.1016/j.amjms.2019.02.003.
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467, doi:10.1016/j.jacbts.2019.02.006.
- Turner, J.D.; Filer, A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr. Opin. Rheumatol. 2015, 27, 175–182, doi:10.1097/BOR.0000000000000148.
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471, doi:10.1136/rmdopen-2017-000471.
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for Fibrotic Diseases: Nearing the Starting Line. Sci. Transl. Med. 2013, 5, 167sr1, doi:10.1126/scitranslmed.3004700.
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040, doi:10.1038/nm.2807.
- Nihlberg, K. Fibroblasts as Matrix Modulating Cells in Asthma and COPD; Department of Experimental Medical Sciences: Lund University, Sweden, 2009; ISBN 9789186253509.
- Kiener, H.P.; Watts, G.F.M.; Cui, Y.; Wright, J.; Thornhill, T.S.; Sköld, M.; Behar, S.M.; Niederreiter, B.; Lu, J.; Cernadas, M.; et al. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ cul-ture. Arthritis Rheum. 2010, 62, 742–752, doi:10.1002/art.27285.
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255, doi:10.1111/j.0105-2896.2009.00859.x.
- Filer, A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr. Opin. Pharmacol. 2013, 13, 413–419, doi:10.1016/j.coph.2013.02.006.
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001, doi:10.1038/nrdp.2018.1.
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219, doi:10.1056/NEJMra1004965.
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33, doi:10.1038/nrrheum.2012.190.
- Juarez, M.; Filer, A.; Buckley, C. Fibroblasts as therapeutic targets in rheumatoid arthritis and cancer. Swiss Med. Wkly. 2012, 142, 1–9, doi:10.4414/smw.2012.13529.
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immu-nol. 2019, 10, 1–8, doi:10.3389/fimmu.2019.01395.
- Hirohata, S.; Yanagida, T.; Nagai, T.; Sawada, T.; Nakamura, H.; Yoshino, S.; Tomita, T.; Ochi, T. Induction of fibroblast-like cells from CD34+ progenitor cells of the bone marrow in rheumatoid arthritis. J. Leukoc. Biol. 2001, 70, 413–421, doi:10.1189/jlb.70.3.413.
- Roelofs, A.J.; Zupan, J.; Riemen, A.H.K.; Kania, K.; Ansboro, S.; White, N.; Clark, S.M.; De Bari, C. Joint morphogenetic cells in the adult mammalian synovium. Nat. Commun. 2017, 8, doi:10.1038/ncomms15040.
- Hardy, R.S.; Hülso, C.; Liu, Y.; Gasparini, S.J.; Fong-Yee, C.; Tu, J.; Stoner, S.; Stewart, P.M.; Raza, K.; Cooper, M.S.; et al. Characterisation of fibroblast-like synoviocytes from a murine model of joint inflammation. Arthritis Res. Ther. 2013, 15, R24, doi:10.1186/ar4158.
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Körner, H.; Wei, W. Ontology and Function of Fibroblast-Like and Macrophage-Like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immu-nol. 2018, 9, doi:10.3389/fimmu.2018.01467.
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789, doi:10.1038/s41467-018-02892-y.
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251, doi:10.1038/s41586-019-1263-7.
- Zerrouk, N.; Miagoux, Q.; Dispot, A.; Elati, M.; Niarakis, A. Identification of putative master regulators in rheumatoid ar-thritis synovial fibroblasts using gene expression data and network inference. Sci. Rep. 2020, 10, 16236, doi:10.1038/s41598-020-73147-4.
- Karami, J.; Aslani, S.; Jamshidi, A.; Garshasbi, M.; Mahmoudi, M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019, 702, 8–16, doi:10.1016/j.gene.2019.03.033.
- Karouzakis, E.; Gay, R.E.; Gay, S.; Neidhart, M. Epigenetic control in rheumatoid arthritis synovial fibroblasts. Nat. Rev. Rheumatol. 2009, 5, 266–272, doi:10.1038/nrrheum.2009.55.
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumor Biol. 2016, 37, 8503–8514, doi:10.1007/s13277-016-5049-3.
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331, doi:10.1016/j.yexcr.2010.02.045.
- Micke, P.; Tman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45, S163–S175, doi:10.1016/j.lungcan.2004.07.977.
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447, doi:10.1002/path.1398.
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337, doi:10.1038/nature03096.
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722, doi:10.1016/j.yexcr.2010.04.032.
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011, doi:10.1186/bcr138.
- Ellis, M.J.C.; Singer, C.; Hornby, A.; Rasmussen, A.; Cullen, K.J. Insulin-like growth factor mediated stromal-epithelial in-teractions in human breast cancer. Breast Cancer Res. Treat. 1994, 31, 249–261, doi:10.1007/BF00666158.
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Repro-gramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed Res. Int. 2018, 2018, 1–12, doi:10.1155/2018/6075403.
- Piek, E.; Heldin, C.-H.; Dijke, P. Ten Specificity, diversity, and regulation in TGF‐β superfamily signaling. FASEB J. 1999, 13, 2105–2124, doi:10.1096/fasebj.13.15.2105.
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114, 4359–4369.
- De Wever, O.; Nguyen, Q.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin‐C and SF/HGF pro-duced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018, doi:10.1096/fj.03-1110fje.
- Grugan, K.D.; Miller, C.G.; Yao, Y.; Michaylira, C.Z.; Ohashi, S.; Klein-Szanto, A.J.; Diehl, J.A.; Herlyn, M.; Han, M.; Nak-agawa, H.; et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcino-ma invasion. Proc. Natl. Acad. Sci. USA 2010, 107, 11026–11031, doi:10.1073/pnas.0914295107.
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453, doi:10.1016/j.molmed.2013.05.004.
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556, doi:10.1073/pnas.1600363113.
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Rønnov-Jessen, L. Epithelial to Mesenchymal Transition in Human Breast Cancer Can Provide a Nonmalignant Stroma. Am. J. Pathol. 2003, 162, 391–402, doi:10.1016/S0002-9440(10)63834-5.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128, doi:10.1158/0008-5472.CAN-07-3127.
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adhes. Migr. 2012, 6, 220–230, doi:10.4161/cam.20875.
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Can-cer Cell 2011, 19, 257–272, doi:10.1016/j.ccr.2011.01.020.
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone Marrow Contribution to Tumor-Associated Myofibroblasts and Fibroblasts. Cancer Res. 2004, 64, 8492–8495, doi:10.1158/0008-5472.CAN-04-1708.
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809, doi:10.1002/pros.20927.
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer‐associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717, doi:10.1111/cas.14537.
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karls-son, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150, doi:10.1038/s41467-018-07582-3.
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Sin-gle-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307, doi:10.3390/cancers12051307.
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351, doi:10.1158/2159-8290.CD-19-1384.
- Du, H.; Che, G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett. 2017, 13, 3–12, doi:10.3892/ol.2016.5451.
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of can-cer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059, doi:10.1016/j.biocel.2012.08.005.
- Enkelmann, A.; Heinzelmann, J.; von Eggeling, F.; Walter, M.; Berndt, A.; Wunderlich, H.; Junker, K. Specific protein and miRNA patterns characterise tumour-associated fibroblasts in bladder cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 751–759, doi:10.1007/s00432-010-0932-6.
- Kekeeva, T.V.; Popova, O.P.; Shegai, P.V.; Alekseev, B.Y.; Andreeva, Y.Y.; Zaletaev, D.V.; Nemtsova, M.V. Aberrant methyl-ation of p16, HIC1, N33, and GSTP1 in tumor epithelium and tumor-associated cells in prostate cancer. Mol. Biol. 2007, 41, 70–76, doi:10.1134/S0026893307010104.
- Nielsen, B.S.; Jørgensen, S.; Fog, J.U.; Søkilde, R.; Christensen, I.J.; Hansen, U.; Brünner, N.; Baker, A.; Møller, S.; Nielsen, H.J. High levels of microRNA-21 in the stroma of colorectal cancers predict short disease-free survival in stage II colon cancer patients. Clin. Exp. Metastasis 2011, 28, 27–38, doi:10.1007/s10585-010-9355-7.
- Luczak, M.W.; Jagodziński, P.P. The role of DNA methylation in cancer development. Folia Histochem. Cytobiol. 2006, 44, 143–154, doi:10.5603/4561.
- Mishra, P.; Kiebish, M.A.; Cullen, J.; Srinivasan, A.; Patterson, A.; Sarangarajan, R.; Narain, N.R.; Dobi, A. Genomic altera-tions of Tenascin C in highly aggressive prostate cancer: A meta-analysis. Genes Cancer 2019, 10, 150–159, doi:10.18632/genesandcancer.196.

