 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ellen Reisinger | + 1517 word(s) | 1517 | 2020-12-07 06:45:12 | | | |
| 2 | Nicole Yin | -28 word(s) | 1489 | 2021-01-13 02:52:27 | | |
Video Upload Options
Otoferlin, an essential synaptic protein in the auditory sensory inner hair cells, is encoded by the gene OTOF. Biallelic variants in OTOF are associated with autosomal recessive auditory neuropathy or synaptopathy (DFNB9). Since its discovery in 1996, roughly 220 causally-associated variants have been uncovered. The prevalence of OTOF-associated hearing loss varies according to population. A higher molecular genetic diagnostic yield has been reported in patients with prelingual auditory synaptopathy, making this very specific clinical feature a useful criterion for OTOF diagnostic testing. Apart from this clinical hallmark, genotype-phenotype correlations are rather complex and include a variant that causes temperature-sensitive auditory neuropathy (p.Ile515Thr) and three variants that have so far been associated with progressive hearing impairment. Several founder variants have been reported in the Asian and European populations. A complete landscape of genomic variation at the DFNB9 locus remains to be determined and improvements in genomics technologies may eventually uncover novel insights into possible missing variants. A complete understanding of OTOF variants is essential for the success of current and future therapies.
1. Introduction
Sensorineural hearing loss is one of the most common sensory deficits in humans, affecting one to two per 1000 newborns in developed countries [1]. Over the past 25 years since the discovery of the first deafness gene, more than 120 genes have been causally associated with non-syndromic hearing loss (https://hereditaryhearingloss.org/) and over 6000 disease-causing variants have been identified [2]. As most variants implicated in hearing loss are small insertions/deletions (indels) or single nucleotide variants [2], high-throughput sequencing is a well-suited method to rapidly allow for a deeper understanding of the spectrum of variants involved in deafness and their consequences on the auditory phenotype.
Using a candidate gene approach, the DFNB9 locus (OMIM: 601071) was mapped to chromosome 2p23.1 in 1996 by studying a genetically isolated family from Lebanon [3]. Three years later, the gene OTOF (OMIM: 603681), encoding a transmembrane (TM) protein called otoferlin, was mapped to the DFNB9 locus and identified as causing prelingual autosomal recessive, non-syndromic deafness [4]. Biallelic pathogenic variants in OTOF cause auditory synaptopathy due to deficient pre-synaptic neurotransmitter release at the ribbon synapse of the inner hair cells (IHCs) [5]. Since its initial identification, about 220 pathogenic and likely pathogenic variants in OTOF have
been identified.
2. Molecular Epidemiology of OTOF-Associated Hearing Loss
2.1. Summary of Variants Identified in Otoferlin
By virtue of being one of the first deafness genes identified, OTOF has been tested in molecular genetic diagnostic settings for over two decades, allowing an estimate of the global burden of OTOF-associated hearing loss. There are presently 219 genetic changes that are classified as pathogenic or likely pathogenic according to the literature or clinical database entries (Leiden Open Variation Database v3.0 (LOVD v3), the Deafness Variation Database (DVD), ClinVar, and the Human Gene Mutation Database (HGMD)). This includes 84 missense, 44 frameshift, 43 nonsense, 36 splice site, 7 in-frame duplications or deletions, 3 copy number variations, as well as 1 stop loss and regulatory variant each (Figures 1 and 2).
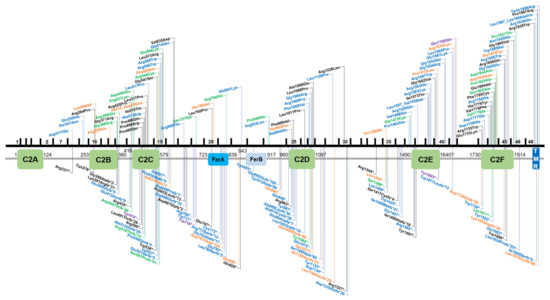
Figure 1. Overview of OTOF variants that are classified as pathogenic/likely pathogenic in the databases ClinVar, Leiden Open Variation Database v3.0 (LOVD v3), the Deafness Variation Database (DVD) or Human Gene Mutation Database (HGMD). Variants in the upper part of the figure are non-truncating, variants below are truncating. Black text indicates homozygous variants, blue and green text represents compound heterozygous and heterozygous variants, respectively. Orange text show variants that are reported in databases without a publication reference with undetermined zygosity. Purple text indicates two different variants on the cDNA level that cause the same protein-level change. Variants are annotated according to NM_001287489.1, encoding NP_001274418.1, or isoform e.
Figure 2. Overview of OTOF splice variants according to ClinVar, Leiden Open Variation Database v3.0 (LOVD v3), the Deafness Variation Database (DVD) or the Human Gene Mutation Database (HGMD). Black text indicates homozygous variants, blue and green text represents compound heterozygous and heterozygous variants, respectively. Orange text shows variants that are reported in databases without a publication reference with undetermined zygosity. Variants are annotated according to NM_001287489.1, encoding NP_001274418.1, or isoform e.
2.2. Population-Based Diagnostic Rates of Otoferlin
The prevalence of OTOF-associated hearing loss varies according to population background. For example, OTOF variants account for approximately 5% of genetic diagnoses in the Turkish population [6], and 3.1% of diagnoses in the Pakistani population [32]. A common founder variant (p.Gln829*) was identified in 3% of Spanish cohorts [7][8]. In other populations, OTOF has been identified as a cause of hearing impairment in 3.1% of Taiwanese [9], 2.4% (primarily) European-American [10], 2–3% of Pakistani [11][12], 1.9% of French [13] and 1.7% of Japanese [14] patients who were not pre-selected on the basis of auditory neuropathy/synaptopathy. In Iranian patients, a study that included 38 consanguineous patients identified only one family with a homozygous frameshift variant (c.1981dupG, p.Asp661Glyfs*2) and suggested OTOF is not a major contributor to hearing loss in the Iranian population [15].
2.3. Diagnostic Rates of Otoferlin in Patients with Auditory Neuropathy/Synaptopathy
Auditory synaptopathy with prelingual onset has been identified in patients with genetic aberrations in a small subset of genes (PJVK, OPA1, and DIAPH3 (AUNA1 locus)), and a limited number of suspected cases in a few other genes such as GJB2 [16][17][18][19][20][21], although the GJB2 cases are controversially discussed [22]. The unique phenotypic presentation of DFNB9 makes a targeted selection for OTOF screening in patients for genetic testing rather successful. As exemplified by a study that included Japanese patients with auditory neuropathy/synaptopathy, biallelic OTOF variants were uncovered in 56% of cases that included the identification of a founder variant (p.Arg1939Gln) [23]. The p.Gln829* founder variant was identified in 87% of patients diagnosed with auditory neuropathy/synaptopathy in the Spanish population [7]. Another founder variant (p.Glu1700Gln) in Taiwanese patients with progressive, moderate-to-profound hearing loss was identified that diagnosed 23% of a selected patient cohort of 22 individuals with auditory neuropathy/synaptopathy [24]. A study that screened the OTOF gene in 37 Chinese patients with congenital auditory neuropathy/synaptopathy had a diagnostic yield of 41.2% [25]. On the contrary, a study that involved the screening of 73 Chinese Han patients with auditory neuropathy/synaptopathy resolved only 5.5% of patients and uncovered a temperature-sensitive variant, which was lower than anticipated and demonstrates a high diagnostic variability [26].
2.4. Missing Variants
The diagnostic yield of patients with audiological hallmarks of DFNB9 suggests multifaceted deficits in general isoform and variant knowledge, as well as possible technical limitations. Beyond the possibility of additional genes harboring causally associated variants that evoke the same clinical features, there are several reasons explaining why patients with auditory synaptopathy due to biallelic variants in OTOF remain undiagnosed after molecular genetic screening. Such reasons include possible limitations stemming from methodology (e.g., sequencing coverage gaps), missed copy number variations that either fall below the detection resolution of commonly used microarrays in genetic diagnostics or missed due to uneven high-throughput sequencing coverage, especially in the case of exome sequencing, or deep intronic variants that are not captured in targeted enrichment approaches. Furthermore, variant interpretation bottlenecks that could also be due to incorrect transcript usage in variant annotation, current limitations in knowledge about the pathogenicity of rare variants and lack of opportunity for segregation testing in families that can complicate outcomes for definitive statements about variant pathogenicity. Another hypothesis points to variants occurring in currently unannotated exons.
Sequence analysis is primarily focused on exonic regions and relies on the complete understanding of gene isoform structure (i.e., exon annotation). The cochlea is encased in one of the hardest bones of the body, making it one of the least accessible tissues for transcriptome studies. However, many microarray and RNA-seq-based studies using the human and rodent whole cochlea have ensued since the early 2000s [27][28]. Though challenging, single-cell isolation of the inner ear and long read single-cell RNA-seq have recently been performed in mice at several developmental time points [29] to reveal cell-type defining genes and pathways. Long-read sequencing and isoform analysis has identified unappreciated splicing heterogeneity and expression of cell-specific isoforms with unannotated exons [29]. A recent study marked a crucial gap in this understanding in many well-studied genes, such as Otof, by mapping a novel non-coding exon 6b and suggesting an in-frame exon 10b (Figure 3). Extending this finding by annotating novel OTOF exons in humans could yield significant implications for undiagnosed patients who would otherwise fit the characteristic DFNB9 phenotypic spectrum.
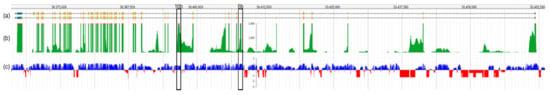
Figure 3. Overview of the Otof transcript structure based on single-cell RNA-seq data from mouse IHCs. (a) Transcript structure of Otof, with exon 1 on the right side of the figure. Exons are depicted as gold bars. Ensembl transcripts Otof-201 (upper isoform, encoding 1997 amino acids) and Otof-202 (lower isoform, encoding 1992 amino acids) are shown. (b) The read depth of each exon is shown in green, with the highest covered regions showing regions in red. Note that maximum read peaks (red) correspond with exons shown in (a) if they are expressed in the IHC. (c) Mammalian conservation track for Otof. Conserved sequences are shown in blue and those not conserved are shown in red. The predicted novel exons 6b and 10b are marked with black boxes. This figure was generated by querying morlscrnaseq.org using the “Transcript Structure Browser” tool [29].
References
- Cynthia C. Morton; Walter E. Nance; Newborn Hearing Screening — A Silent Revolution. New England Journal of Medicine 2006, 354, 2151-2164, 10.1056/nejmra050700.
- Hela Azaiez; Kevin T. Booth; Sean S. Ephraim; Bradley Crone; Elizabeth A. Black-Ziegelbein; Robert J. Marini; A. Eliot Shearer; Christina M. Sloan-Heggen; Diana Kolbe; Thomas Casavant; et al.Michael J. SchniedersCarla NishimuraTerry BraunRichard J.H. Smith Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. The American Journal of Human Genetics 2018, 103, 484-497, 10.1016/j.ajhg.2018.08.006.
- Hassan Chaïb; Christophe Place; Nabiha Salem; Sébastien Chardenoux; Christophe Vincent; Jean Weissenbach; Elie El-Zir; Jacques Loiselet; Christine Petit; A Gene Responsible for a Sensorineural Nonsyndromic Recessive Deafness Maps to Chromosome 2p22-23. Human Molecular Genetics 1996, 5, 155-158, 10.1093/hmg/5.1.155.
- Shin'ichiro Yasunaga; M'hamed Grati; Martine Cohen-Salmon; Aziz El-Amraoui; Mirna Mustapha; Nabiha Salem; Elie El-Zir; Jacques Loiselet; Christine Petit; A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nature Genetics 1999, 21, 363-369, 10.1038/7693.
- Isabelle Roux; Saaid Safieddine; Régis Nouvian; M'hamed Grati; Marie-Christine Simmler; Amel Bahloul; Isabelle Perfettini; Morgane Le Gall; Philippe Rostaing; Ghislaine Hamard; et al.Antoine TrillerPaul AvanTobias MoserChristine Petit Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell 2006, 127, 277-289, 10.1016/j.cell.2006.08.040.
- Duygu Duman; Asli Sirmaci; F. Basak Cengiz; Hilal Ozdag; Mustafa Tekin; Screening of 38 Genes Identifies Mutations in 62% of Families with Nonsyndromic Deafness in Turkey. Genetic Testing and Molecular Biomarkers 2011, 15, 29-33, 10.1089/gtmb.2010.0120.
- Montserrat Rodríguez-Ballesteros; Raúl Reynoso; Margarita Olarte; Manuela Villamar; Constantino Morera; Rosamaria Santarelli; Edoardo Arslan; Carme Medá; Carlos Curet; Christiane Völter; et al.Manuel Sainz-QuevedoPierangela CastorinaUmberto AmbrosettiStefano BerrettiniKlemens FreiSocorro TedínJanine SmithM. Cruz TapiaLaura CavalléNancy GelvezPaola PrimignaniElena Gómez-RosasMirta MartínMiguel A. Moreno-PelayoMartalucía TamayoJosé Moreno-BarralFelipe MorenoIgnacio Del Castillo A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Human Mutation 2008, 29, 823-831, 10.1002/humu.20708.
- V Migliosi; S Modamio-Høybjør; M A Moreno-Pelayo; M Rodríguez-Ballesteros; M Villamar; D Tellería; I Menéndez; F Moreno; I Del Castillo; Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. Journal of Medical Genetics 2002, 39, 502-506, 10.1136/jmg.39.7.502.
- Wu; Cheng-Yu Tsai; Lin; Chen; Yen-Fu Cheng; Chee-Yee Lee; Jargalkhuu Erdenechuluun; Tien-Chen Liu; Chuan-Jen Hsu; Tsai; et al.LeeLiuHsuChen-Chi WuYi-Hsin LinPey-Yu ChenPei-Hsuan LinChe-Ming WuYin-Hung LinPei-Lung Chen Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772, 10.3390/genes10100772.
- Christina M. Sloan-Heggen; Amanda O. Bierer; A. Eliot Shearer; Diana L. Kolbe; Carla J. Nishimura; Kathy L. Frees; Sean S. Ephraim; Seiji B. Shibata; Kevin T. Booth; Colleen A. Campbell; et al.Paul T. RanumAmy E. WeaverE. Ann Black-ZiegelbeinDonghong WangHela AzaiezRichard J.H. Smith Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Quality of Life Research 2016, 135, 441-450, 10.1007/s00439-016-1648-8.
- B Y Choi; Z M Ahmed; S Riazuddin; M A Bhinder; M Shahzad; T Husnain; A J Griffith; Thomas B. Friedman; Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clinical Genetics 2009, 75, 237-243, 10.1111/j.1399-0004.2008.01128.x.
- Elodie M. Richard; Regie Lyn P. Santos-Cortez; Rabia Faridi; Atteeq U. Rehman; K. Lee; Mohsin Shahzad; Anushree Acharya; Asma A. Khan; Ayesha Imtiaz; Imen Chakchouk; et al.Christina TaklaIzoduwa AbbeMaria RafeeqKhurram LiaqatTaimur ChaudhryMichael J. BamshadDebbie NickersonIsabelle SchrauwenShaheen N KhanRobert J. MorellSaba ZafarMuhammad AnsarZubair M AhmedW. AhmadSheikh RiazuddinThomas B. FriedmanSuzanne M. LealSaima RiazuddinUniversity of Washington Center for Mendelian GenomicsRegie Lp. Santos-CortezSheik Riazuddin Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Human Mutation 2018, 40, 53-72, 10.1002/humu.23666.
- David Baux; C. Vaché; C. Blanchet; M. Willems; C. Baudoin; M. Moclyn; V. Faugère; R. Touraine; B. Isidor; D. Dupin-Deguine; et al.M. NizonM. VincentS. MercierC. CalaisG. García-GarcíaZ. AzherL. LambertY. Perdomo-TrujilloF. GiulianoM. ClaustresM. KoenigM. MondainAnne‐Françoise Roux Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Scientific Reports 2017, 7, 16783, 10.1038/s41598-017-16846-9.
- Yoh-Ichiro Iwasa; Shin-Ya Nishio; Akiko Sugaya; Yuko Kataoka; Yukihiko Kanda; Mirei Taniguchi; Kyoko Nagai; Yasushi Naito; Tetsuo Ikezono; Rie Horie; et al.Yuika SakuraiRina MatsuokaHidehiko TakedaSatoko AbeChiharu KiharaTakashi IshinoShin-Ya MoritaSatoshi IwasakiMasahiro TakahashiTsukasa ItoYasuhiro AraiShin-Ichi Usami OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLOS ONE 2019, 14, e0215932, 10.1371/journal.pone.0215932.
- Nejat Mahdieh; Atefeh Shirkavand; Bahareh Rabbani; Mustafa Tekin; Bahman Akbari; Mohammad Taghi Akbari; Sirous Zeinali; Screening of OTOF mutations in Iran: A novel mutation and review. International Journal of Pediatric Otorhinolaryngology 2012, 76, 1610-1615, 10.1016/j.ijporl.2012.07.030.
- Sedigheh Delmaghani; Francisco J. Del Castillo; Vincent Michel; Michel Leibovici; Asadollah Aghaie; Uri Ron; Lut Van Laer; Nir Ben-Tal; Guy Van Camp; Dominique Weil; et al.Francina LangaMark LathropPaul AvanChristine Petit Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nature Genetics 2006, 38, 770-778, 10.1038/ng1829.
- Patrizia Amati-Bonneau; Agnès Guichet; Aurelien Olichon; Arnaud Chevrollier; Frederique Viala; Stéphanie Miot; Carmen Ayuso; Sylvie Odent; Catherine Arrouet; Christophe Verny; et al.Marie-Noelle CalmelsGilles SimardPascale BelenguerJing WangJ.L PuelChristian HamelYves MalthièryMinique BonneauGuy LenaersPascal Reynier OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Annals of Neurology 2005, 58, 958-963, 10.1002/ana.20681.
- Arnold Starr; Brandon Isaacson; Henry J. Michalewski; Fan-Gang Zeng; Ying-Yee Kong; Paula Beale; George W. Paulson; Bronya J.B. Keats; Marci M. Lesperance; A Dominantly Inherited Progressive Deafness Affecting Distal Auditory Nerve and Hair Cells. Journal of the Association for Research in Otolaryngology 2004, 5, 411-426, 10.1007/s10162-004-5014-5.
- T B Kim; B Isaacson; T A Sivakumaran; A Starr; B J B Keats; M M Lesperance; A gene responsible for autosomal dominant auditory neuropathy (AUNA1) maps to 13q14-21. Journal of Medical Genetics 2004, 41, 872-876, 10.1136/jmg.2004.020628.
- Xing Cheng; Li Li; Shanda Brashears; Thierry Morlet; San San Ng; Charles Berlin; Linda Hood; Bronya J B Keats; Connexin 26 variants and auditory neuropathy/dys-synchrony among children in schools for the deaf. American Journal of Medical Genetics Part A 2005, 139, 13-18, 10.1002/ajmg.a.30929.
- Rosamaria Santarelli; Elona Cama; Pietro Scimemi; Erica Dal Monte; Elisabetta Genovese; Edoardo Arslan; Audiological and electrocochleography findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. European Archives of Oto-Rhino-Laryngology 2007, 265, 43-51, 10.1007/s00405-007-0412-z.
- Del Castillo Francisco J.; Genetics of isolated auditory neuropathies. Frontiers in Bioscience 2012, 17, 1251, 10.2741/3984.
- Tatsuo Matsunaga; H Mutai; S Kunishima; K Namba; N Morimoto; Y Shinjo; Y Arimoto; Y Kataoka; T Shintani; N Morita; et al.T SugiuchiS MasudaA NakanoH TaijiK Kaga A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clinical Genetics 2012, 82, 425-432, 10.1111/j.1399-0004.2012.01897.x.
- Yu-Hsun Chiu; Chen-Chi Wu; Ying-Chang Lu; Pei-Jer Chen; Wen-Yuan Lee; Alyssa Yan-Zhen Liu; Chuan-Jen Hsu; Mutations in the OTOF Gene in Taiwanese Patients with Auditory Neuropathy. Audiology and Neurotology 2010, 15, 364-374, 10.1159/000293992.
- Qiu‐Jing Zhang; Bing Han; Lan Lan; Liang Zong; Wei Shi; Hong‐Yang Wang; Lin‐Yi Xie; Cui Zhao; Zi‐Fang Yin; Christine Petit; et al.Jing Guan High frequency of OTOF mutations in Chinese infants with congenital auditory neuropathy spectrum disorder. Clinical Genetics 2016, 90, 238-246, 10.1111/cge.12744.
- Dayong Wang; Yi-Chen Wang; Dominique Weil; Yali Zhao; Shao-Qi Rao; Liang Zong; Yu-Bin Ji; Qiong Liu; Jian-Qiang Li; Huangming Yang; et al.Y. ShenCindy Benedict-AlderferQing Yin ZhengChristine PetitQiuju Wang Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Medical Genetics 2010, 11, 79-79, 10.1186/1471-2350-11-79.
- Younsook Cho; Tzy-Wen L. Gong; Margaret I. Lomax; Richard A. Altschuler; Timo Stöver; Gene Expression Profiles of the Rat Cochlea, Cochlear Nucleus, and Inferior Colliculus. Journal of the Association for Research in Otolaryngology 2002, 3, 54-67, 10.1007/s101620010042.
- Tiantian Cai; Hsin-I Jen; Hyojin Kang; Tiemo J. Klisch; Huda Y. Zoghbi; Andrew K. Groves; Characterization of the Transcriptome of Nascent Hair Cells and Identification of Direct Targets of the Atoh1 Transcription Factor. The Journal of Neuroscience 2015, 35, 5870-5883, 10.1523/jneurosci.5083-14.2015.
- Paul T. Ranum; Alexander T. Goodwin; Hidekane Yoshimura; Diana L. Kolbe; William D. Walls; Jin-Young Koh; David Z.Z. He; Richard J.H. Smith; Insights into the Biology of Hearing and Deafness Revealed by Single-Cell RNA Sequencing. Cell Reports 2019, 26, 3160-3171.e3, 10.1016/j.celrep.2019.02.053.

