 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Velia Siciliano | + 3838 word(s) | 3838 | 2021-01-12 11:00:49 | | | |
| 2 | Peter Tang | -99 word(s) | 3739 | 2021-01-17 09:15:31 | | |
Video Upload Options
Engineered mammalian cells for medical purposes are becoming a clinically relevant reality thanks to advances in synthetic biology that allow enhanced reliability and safety of cell-based therapies. However, their application is still hampered by challenges including time-consuming design-and-test cycle iterations and costs. For example, in the field of cancer immunotherapy, CAR-T cells targeting CD19 have already been clinically approved to treat several types of leukemia, but their use in the context of solid tumors is still quite inefficient, with additional issues related to the adequate quality control for clinical use.
1. Introduction
Cancer immunotherapy has the goal of improving anti-tumor immune responses reducing off-target effects typical of chemotherapies and other state-of-the-art treatments. Since cancer cells often evade the surveillance of the immune system, immunotherapies have the function of priming the immune response to make it more efficient. Different classes of immunotherapies have already been approved for cancer treatment and some others are in clinical trials [1] intending to facilitate the recognition of cancer cells by the immune system [2][3]. These include checkpoint inhibitors, lymphocyte activating cytokines, agonists for co-stimulatory receptors, cancer vaccines, oncolytic viruses, bispecific antibodies and T cell-based adoptive immunotherapy (ACT) [2][3]. Checkpoint inhibitors are the most relevant and largely studied immunotherapeutic drugs up to date. They act by blocking co-inhibitory molecules binding to their cognate ligands on the surface of cancer cells. The two most common strategies are the PD-1/PD-L1 axis blockade and the inhibition of CTLA-4, to prolong T cell activity and anti-tumoral effects [4]. A limitation of immune checkpoint inhibitors is that they can cause immune-related adverse events (irAEs), usually related to autoimmunity in a dose-independent manner [5][6]. In addition, many patients do not respond to this therapy due to the low number of tumor-infiltrating lymphocytes (TILs), downregulation of co-inhibitory molecules in both tumor cells and T cells, and adapted resistance [4][7].
Cytokines act by actively limiting tumor growth with a direct anti-proliferative or pro-apoptotic action or by enhancing tumor recognition and cytotoxicity of the immune system against cancer cells [8]. Nevertheless, several issues including the pleiotropic function of many cytokines, which can act both as immunosuppressors or activators depending on the cellular context, the redundancy of cytokine signaling, and the short half-life of these molecules make the efficacy of these treatments difficult. This therapy often consists of the administration of high doses of cytokines that can cause vascular leakage and cytokine release syndrome (CRS) [9]. A recent strategy, still at an earlier phase of development, is the use of agonist antibodies designed to specifically bind co-stimulatory molecules, such as CD28 or the inducible T cell co-stimulator (ICOS), increasing T cell proliferation and survival in the immunoglobulin-like superfamily, as well as OX40, CD27, and 4-1BB in the TNFR superfamily [10]. Of note, agonist antibodies are designed to bind and activate the target receptor, mimicking the action of the natural ligand, and the kinetics of this binding may depend on many factors, such as the affinity to the epitope, and the interaction between the antibody fragment crystallizable domain (Fc) and its receptor.
Lastly, cancer vaccines represent another promising strategy to increase the immune response against cancer. They are divided into (i) cell vaccines, obtained from the tumor or immune cells, (ii) protein/peptides vaccines based on tumor-associated antigens (TAAs), and (iii) genetic vaccines, which use viral, plasmid vectors, or mRNA from autologous tumor tissue to deliver the antigen.
Cancer vaccines can be either prophylactic or therapeutic. Examples of prophylactic vaccines include the one against hepatitis B virus which accounts for liver cancer, and the one against human papillomavirus, responsible for the most cervical cancers [11].
Therapeutic vaccines have the goal to increase the tumor-specific adaptive response. Cancer vaccines have shown reduced toxicity and autoimmunity issues, although their efficacy is, for now, lower than other immunotherapeutic strategies, due to (i) an inappropriate activation of effector cells, (ii) limited accumulation of these cells in the tumor, and (iii) to the immunosuppressive tumor microenvironment. Advances in synthetic biology allowed engineering safer and more effective vectors, DNA or RNA–based implemented on bacterial or viral backbones, carrying genetic components, and functional resources [12].
2. T Cell-Based Therapies
T cells have a prominent key role in the immune response against cancer. TILs are commonly found in the tumor microenvironment (TME), where they can exert anti-tumor actions [13]. Their presence is frequently associated with a better prognosis, even though a cytotoxic subset such as CD8+ T cells may undergo impaired activation in the TME. The isolation of these cells from excised tumors and the subsequent reinvigoration in vitro before reintroducing them back to the patient is one of the strategies currently used in clinics to increase T cells response against tumor [13].
Leveraging on immune cell infiltration in the TME, over the past years, several therapies have been developed to improve their action against tumors. In particular, T cell-based adoptive immunotherapy (ACT) is a novel anticancer treatment that consists of the in vitro expansion and activation of autologous immune cells, prior to reinfusion into patients [14]. So far, this treatment has been used mainly for hematologic cancers, although they have been recently tested also for solid tumors [15]. ACTs include (i) tumor-infiltrating lymphocytes (TILs) from an endogenous source, (ii) T cell receptor (TCR)-modified T cells, and (iii) chimeric antigen receptor T cells (CAR-T cell) [16]. The use of TILs for cancer treatment is achieved by harvesting CD4+ and CD8+ T lymphocytes from the patient's tumor, followed by in vitro expansion in a medium supplemented with IL-2 alone or in combination with IL-7, IL-15, and IL-21. TILs are composed of an enriched polyclonal population and are reinfused in the patient to increase the immune response by recognizing tumor-associated antigens as well as tumor neoantigens, and mutated antigens expressed only by tumor cells [17].
TCR-modified T cells are first isolated from tumor patients, and genetically modified with a TCR that is engineered to specifically recognize antigens expressed on cancer cells presented by the MHC system [18][19]. Instead, CAR-T cells are genetically engineered to express a Chimeric Antigen Receptor (CAR) composed of two domains: (1) an extracellular domain consisting of an antibody single-chain fragment (ScFv) that specifically recognizes cell surface antigens on tumor cells and (2) a chimeric intracellular domain formed by the activating intracellular domains from TCR complex (CD3ζ) and other co-stimulatory molecules. Unlike conventional TCR-mediated activation, CAR-mediated activation is antigen-specific and MHC-unrestricted [20][21]. CAR-T has proven higher efficacy, at least against hematological malignancies such as B cell neoplasia and multiple myeloma.
Unfortunately, CAR-T therapy does not exhibit the same efficacy in solid tumors as it does for blood cancers. This is due to several reasons among which tumor heterogeneity and T cell dysfunction are caused by tonic signaling coming from tumor cells and chronic antigen exposure. Additionally, the immunosuppressive features of the tumor microenvironment (i.e., hypoxia), the presence of inhibitory myeloid-derived cells (neutrophils, M2 macrophages, myeloid-derived suppressor cells) and regulatory T cells (Tregs), and the inhibitory role of the extracellular matrix, composed by fibrous proteins, collagen, and hyaluronan [22], have been described as potential immune escape mechanisms of solid tumors in the scientific literature. Moreover, solid tumors induce the formation of aberrant tumor vasculature by producing molecules such as VEGF and other proangiogenic factors, which induce the expression of inhibitory receptors, like PD-1, TIM-3, and IDO-1 on relevant cytotoxic immune cell subsets [23].
All these different features have contributed to classifying tumors in cold and hot tumors depending on the amount and relative quantities of lymphocytes at the tumor core and the tumor margin [24]. Hot tumors, that have a high level of infiltrating lymphocytes, represent the best candidates for immune checkpoint inhibition therapies or cell therapies, whereas cold tumors display the lowest response rate [24]. Several approaches have been proposed to increase the response of those tumors to immunotherapy. Among them, combination therapies aiming to enhance T cell responses by removing co-inhibitory signals, such as immune checkpoint inhibitors (ICIs) and myeloid-derived suppressor cells (MDSC) depletion, along with the activation of co-stimulatory signals such as anti-OX40, are currently being developed. Unfortunately, autoimmunity-related issues represent a major limitation when these approaches are implemented in patients [25].
It has been reported that CAR-T therapy exhibits some side effects, including neurotoxicity and Cytokine Release Syndrome (CRS), that to date are not deeply understood. Symptoms of neurotoxicity include among others headache, confusion, aphasia, attention deficit, memory loss, and only in severe cases cerebral edema, that may lead to death. In the vast majority of the cases, their onset is 4–5 days post-infusion, but are usually reversible and solved in 3–8 weeks after CAR-T cell injection. CRS usually occurs in up to 22 days post-injection and it is solved in 60 days. Symptoms include fever, nausea, fatigue, hypotension, and hypoxia. The involved cytokines in CRS following CAR-T cell therapy includes not only effector cytokines such as interferon (IFN)-γ, IL-2, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) but also the cytokines mainly secreted by the monocytes and/or macrophages such as IL-1, IL-6, IL-8, IL-10, IL-12, tumor necrosis factor (TNF)-α and IFN-α. Neurotoxicity and CRS may be considered linked since they are derived from an extreme immune activation due both to CAR-T cells and non-CAR-T cells [26][27].
3. Synthetic Biology Approaches to Boost CAR-T Cell Treatment's Efficacy
3.1. Addressing the Tumor Immune Escape
The heterogeneous expression pattern of target antigens and antigen-negative relapses in long-term follow-ups prompted the need of targeting more than one antigen synergistically. An OR-gate CAR can be used to recognize two different tumor-associated antigens, requiring only one of them to activate cells. More recent strategies consist in the integration in the same CAR construct of two scFv domains separated by a protein linker, thus forming a bi-specific CAR namely "Tandem" CAR (Figure 1) [28][29][30]. CD19/CD20, as well as CD19/CD22 bi-specific CAR-T cells, are being currently tested on clinical trials (NCT04007029, NCT04215016, NCT03919526, NCT04303520) for the treatment of B cell malignancies.
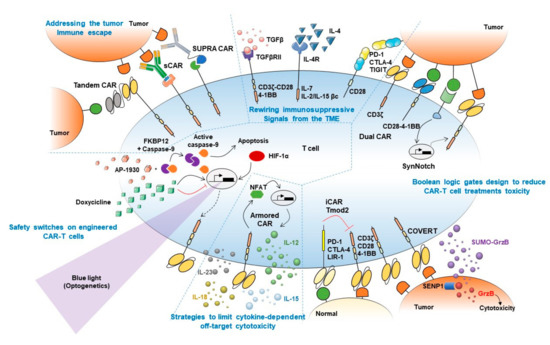
Figure 1. Synthetic biology approaches to boost CAR-T cell treatment's efficacy. Graphical abstract summarizing main synthetic biology approaches developed to prevent current limitations of adoptive T cell therapies. Strategies to boost CAR-T cell therapy efficacy are mechanistically classified into (1) approaches to overcome tumor immune escape (top left), (2) strategies to rewire immunosuppressive signals from the tumor microenvironment (TME) (top middle), (3) Boolean logic gates to reduce CAR-T cell treatment toxicity (right), (4) strategies to limit cytokine-dependent off-target cytotoxicity (bottom middle), and (5) safety switches on engineered CAR-T cells (bottom left). Main strategies to overcome tumor antigen escape depicted in the figure include Tandem (or bi-specific) CAR, switchable CAR or sCAR, and split, universal, and programmable CAR or SUPRA CAR. Fusion proteins combining TGFβ/IL-4 immunosuppressive ectodomains with CD3/CD28-4-1BB or IL-7/IL-2-IL-15 endodomains are included among approaches devised to rewire immunosuppressive signal from the TME. Approaches focused on reducing CAR-T cell treatment toxicity depicted in the figure include Dual CAR, SynNotch, and COVERT strategies, which depend on the presence in target tumor cells of two different antigens to allow T cells activation, and iCAR/Tmod2 approaches that prevent activation of T cells against normal cells that express the antigen recognized by the inhibitory CAR. Strategies to limit cytokine-dependent off-target cytotoxicity include Armored CARs, which can secrete cytokines locally upon activation, preventing systemic undesired side effects induced by repeated systemic cytokines administration. Safety switches included in the figure comprise the iCasp system, which triggers engineered cells apoptosis upon AP-1930 administration, as well as TET-inducible, hypoxia-inducible and light-inducible CAR expression to allow tight control of CAR expression upon doxycycline treatment, hypoxia onset, or blue light stimulation, respectively.
An intelligent approach to obtain a universal and modular platform of T cells to bind several different targets is the peptide-specific switchable CAR-T-cells (sCARs). The general idea consists of an extracellular domain of the CAR to recognize a ligand which is provided as a free molecule fused to an scFv specific to the tumor antigen (Figure 1). Thus the ligand-scFv function as a bridge between the "universal" CAR and the tumor cells, and the cytotoxicity is limited only to cells coated with the antibodies/adapters [31][32][33][34]. An elegant evolution of this strategy is the Split Universal Programmable (SUPRA) CARs [35]. Similar to sCARs, SUPRA CARs are universal CARs composed of a leucine zipper that binds a homologous domain fused to an scFv, creating a synapsis between the CAR and the tumor cells (Figure 1). This design allows fine-tuning of T cell activation through the binding affinity of the leucine zippers, by adding competing leucine zippers and playing with the binding affinity of the scFv.
Overall, these two modular strategies could substantially reduce the costs and time of T cell expansion into antigen-specific CAR-T cells. Furthermore, by adding or withholding the scFv part of the construct it might be able to tightly control CAR-T cells activation, improving CAR-T cells safety.
3.2. Rewiring Immunosuppressive Signals from the TME
It is well known that the TME releases several signals which depress the immune response against cancer. For example, TGFβ can induce a shift of cytotoxic T cells towards a Treg-like phenotype with highly immunosuppressive functions in the TME. To counteract this effect, CAR, where the extracellular part of the TGFβRII is fused to the endodomain of 4-1BB or that link a TGFβ-specific scFv to the CD28-CD3ζ intracellular signaling domains, have been engineered (Figure 1), activating rather than inhibiting CAR-T cells by TGFβ stimulation [36][37]. The ability to convert an immunosuppressive stimulus coming from the TME into an immunostimulatory response was achieved also by fusing the IL-4 receptor ectodomain (an immunosuppressive cytokine) to the IL-7 receptor endodomain or the βc receptor subunit common to IL-2 and IL-15 signaling (immunostimulatory cytokines) (Figure 1) [38][39]. Of note, an undesired limitation of these approaches is that rewiring an immunosuppressive stimulus to an activating/co-stimulatory signal might lower T cells activation threshold, thus favoring OFF-target cytotoxicity onset.
Switch receptor combining immune checkpoint proteins ectodomain fused to co-stimulatory proteins endodomains also showed to enhance T cells antitumor immunity. For example, PD-1-CD28 and CTLA-4-CD28 chimeric receptors demonstrated to boost tumor-specific T cells killing of tumor cells [40][41]. Similarly, T cell immunoreceptor with Ig and ITIM domains (TIGIT) modified following the same strategy, showed better control of tumor growth in vivo (Figure 1) [42]. The engagement of endogenous non-engineered T cells by secretion of bispecific T-cell engagers (BiTEs) is a more recent development. In a preliminary approach, these bi-specific antibodies were secreted by engineered cells and consisted of one scFv targeting EGFR, overexpressed by glioblastoma cells, and the scFv on the other side of the molecule targeting CD3. This strategy facilitated the elimination of tumor xenografts in vivo [43].
3.3. Boolean Logic Gates Design to Reduce CAR-T Cell Treatments Toxicity
By enhancing multiple signals-derived specificities, Boolean logic gates have been demonstrated to be useful to prevent or reduce toxicity concerns arising from immune cellular therapies. By this approach, CAR-T cells would exert their function only in the presence of the two antigens A and B (AND gate) or if the downstream activation of A is blocked by the presence of inhibitory B (AND NOT gate) (Figure 2) [44][45].
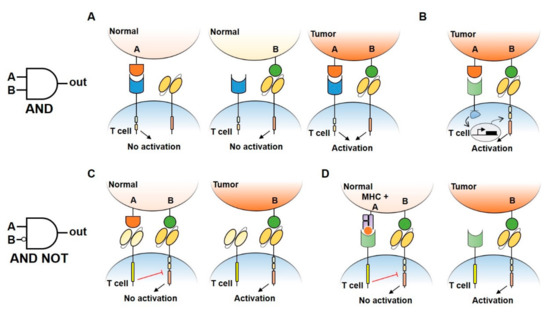
Figure 2. Logic gates design to reduce CAR-T cell treatment toxicity. (A) An AND gate CAR-T design consisting of splitted intracellular activation (CD3ζ signaling domain in the CAR receptor and CD28/4–1BB costimulatory domains in another receptor). Single antigen recognition is not able to activate T cell response, preventing damage in normal tissues. Double antigen recognition (A AND B) drives the activation of T cells and killing of the tumor. (B) An AND gate approach using a synNotch receptor combined with a CAR receptor to increase antigen specificity and safety. Under the recognition of a first antigen by synNotch receptor, an intracellular transcription factor (TF) is released and induces the activation of CAR receptor specific to a second antigen, allowing the activation of T cells. (C,D) By using inhibitory CAR receptors (iCAR) (C) or blocking Tmod2 (D), AND NOT gates allow the engineered cells to distinguish normal tissues by preventing T cell activation when a double antigen recognition is present.
Several strategies that implement AND logic gates on engineered T cells have been developed up to date. One model consists of splitting intracellular activation domains into two different CARs recognizing diverse antigens (e.g., CD3ζ signaling domain in one CAR and CD28/4–1BB costimulatory domains in the other, Figure 2A) [44][46]. Similarly, SynNotch CAR-T cells exert their cytotoxic activity only if two different antigens are present on the target cell surface [47]. In this two-steps approach, the binding of the SynNotch receptor to the first TAA triggers the release of an intracellular transcription factor, which drives the expression of a CAR specific to a second TAA. CAR binding to the second TAA will ultimately promote T cell activation against cells expressing both TAA (Figure 2B). Lastly, an AND-like gate is the Cytoplasmic Oncoprotein VErifier and Response Trigger (COVERT) system. Here, T cells bind the TAA to release a conditionally active cytotoxic protein SUMO-GranzymeB (SUMO-GrzB). GrzB is inactive unless SUMO is removed by the SENP1 protease which is selectively overexpressed by tumor cells. Thus, only in cancer cells, the cleavage of SUMO by SENP1 allows GrzB to unleash its cytotoxic capacity [48].
To limit OFF-side effects of CAR-T, AND NOT logic gates have also been implemented. The co-expression on CAR-T cells of activating and inactivating CARs (iCAR) have been shown to prevent T cell activation against cells expressing two antigens while killing the ones expressing only the antigen recognized by activating CARs. iCAR design consists of a ligand-binding extracellular domain fused to an immune checkpoint protein endodomain (e.g., PD-1, CTLA-4), thus transmitting inhibitory signals upon antigen encounter, and preventing CAR-T cells activation against cells expressing two TAA (Figure 2C). Specifically, T cells were engineered to co-express a second-generation anti-CD19 CAR along with an iCAR targeting the prostate-specific membrane antigen (PSMA). Engineered iCAR-T cells were effectively activated when in contact with cells expressing only CD19 AND NOT PSMA [45]. In line with this work, Hamburguer et al. recently built-up an optimized iCAR system, namely the Tmod2 system [49]. Here, Tmod2 cells are engineered to co-express an activator CAR or TCR recognizing a TAA expressed by tumor and normal cells, as well as a blocker receptor able to recognize a surface antigen that is lost in tumor cells due to loss of heterozygosity (LOH) (Figure 2D). Thus, engineered cells are tuned ON only if the second antigen is absent from the target cell surface. In this system, signal integration relies on the presence of immunoreceptor tyrosine-based activation motif (ITAM) sequences on activating CARs/TCRs and an immunoreceptor tyrosine-based inhibitory motif (ITIM) segment that mediates blocker receptor inhibitory signaling. This seminal study opens the field of targeting missing antigens, rather than the expressed ones, for cancer immunotherapy.
3.4. Strategies to Limit Cytokine-Dependent Off-Target Cytotoxicity
Standard TIL treatment protocols usually require treating patients with repeated high doses of IL-2 which however can also induce the expansion of immunosuppressive Tregs and trigger severe side effects on treated patients [8][50]. Hence, several approaches have been developed to deliver cytokine signaling in engineered T cells, avoiding off-target stimulation (e.g., in Tregs). One of the most original approaches consists of the development of orthogonal IL-2 receptor pairs that are activated by mutated IL-2, which is not able to activate native IL-2 receptors. Thus, with this method, it should be possible to specifically activate engineered T cells avoiding IL-2 OFF-target deleterious effects. This strategy showed to enhance engineered T cells expansion and anti-tumor efficacy, upon exogenous administration of mutant IL-2, in preclinical models [51]. A similar strategy was followed in the engineering of T cells to express constitutively activated IL-7 receptors, thus providing a pro-survival stimulus to engineered cells that leads to enhanced antitumor activity and an increase in T cells persistence in mice [52].
Other approaches trying to confine cytokine signaling to engineered cells involve IL-12 expression. For instance, stably transduced T cells to express IL-12 driven by NFAT synthetic promoter [53] allow IL-12 production upon TCR stimulation of engineered cells. Although validated in preclinical models, this approach led to significant toxicities in clinical trials, which were associated with promoter leakiness [54]. To address this disappointing result, genetically engineered cells with an activation-induced, membrane-anchored version of IL-12 were generated, limiting diffusion of IL-12 and minimizing off-target effects [55]. Tumor-associated T cells expressing IL-18 upon activation were also proved safe and to induce better tumor clearance in preclinical models [56].
This strategy has been also developed for CAR-T cell models. Multiple cytokine-producing CARs, namely "armored" CAR-T cells (Figure 1), including IL-12, IL-15, and IL-18 [57][58][59] are nowadays at early phases of development. Interestingly, IL-23-secreting CAR-T cells stand out for showing better antitumor efficacy and safety profiles than either, IL-15- and IL-18-secreting CAR-T cells [60].
3.5. Safety Switches on Engineered CAR-T Cells
As CAR-T cell therapies can induce a large array of severe, life-threatening side effects in patients, tight spatio-temporal control of CAR-T cell viability represents a fundamental objective to make these therapies safer [43][61][62][63][64]. CAR-T cells were engineered to express modified human caspase-9 fused to human FKBP12 (iCasp9 system, Figure 1) such that exogenous administration of AP1930 (small molecule) induces dimerization of FKBP12 and activation of caspase-9, triggering cell apoptosis [65][66]. Another approach to selectively kill CAR-T cells consist of the co-expression of CD20/tEGFR (truncated EGFR), which can be targeted by Rituximab/Cetuximab. Thus, binding of these antibodies to CAR-T cells trigger antibody-dependent cellular cytotoxicity (ADCC) and subsequently killing of engineered cells [67][68][69].
The counterpart of these strategies is irreversibility. Alternatively, more dynamic ON-OFF strategies were developed to temporarily inactivate engineered T cells in cases of severe side effects. To achieve this, TET-ON and TET-OFF systems (Figure 1) have demonstrated to induce/prevent CAR expression on engineered cells by the addition of tetracycline [71][72]. Moreover, inducible promoters responsive to hypoxia-inducible factor (HIF)-1α showed to induce CAR expression only under hypoxic conditions (Figure 1), thus limiting CAR-T cells activation to hypoxic environments, like the tumor cores [73].
Lastly, optogenetics provides a means of tight spatio-temporal control of CAR-T cell activation. Briefly, optogenetics are based on the engineering of target cells with light-inducible sensors which, upon activation, can trigger the expression of actuator proteins in a tight spatio-temporal manner. By using optogenetic devices based on CRY2 (cryptochrome 2)–CIB1 (cryptochrome-interacting basic-helix-loop-helix 1) system, fused to transcription factors involved in target gene expression, scientists successfully induced IL-2, IL-15, and TNFα production [70], as well as CAR expression [74][75] in engineered T cells upon blue-light stimulation, with no relevant leakiness or constitutive activation of the systems, observed so far (Figure 1). The main limitation of this approach is the scarce penetration of visible light into tissues, which might be overcome by using a different kind of waves able to penetrate deeper into tissues (e.g., ultrasounds).
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34, doi:10.1056/NEJMoa1504030.
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122, doi:10.1158/2326-6066.CIR-15-0190.
- Schmidt, C. The benefits of immunotherapy combinations. Nature 2017, 552, S67–S69, doi:10.1038/d41586-017-08702-7.
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196, doi:10.1038/s41573-018-0006-z.
- Wang, P.-F.; Chen, Y.; Song, S.-Y.; Wang, T.-J.; Ji, W.-J.; Li, S.-W.; Liu, N.; Yan, C.-X. Immune-Related Adverse Events Associated with Anti-PD-1/PD-L1 Treatment for Malignancies: A Meta-Analysis. Front. Pharmacol. 2017, 8,1-20, doi:10.3389/fphar.2017.00730.
- Chen, T.W.; Razak, A.R.; Bedard, P.L.; Siu, L.L.; Hansen, A.R. A systematic review of immune-related adverse event reporting in clinical trials of immune checkpoint inhibitors. Ann. Oncol. 2015, 26, 1824–1829, doi:10.1093/annonc/mdv182.
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547, doi:10.1038/nm.4321.
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15, doi:10.1038/s41416-018-0328-y.
- Lee, S.; Margolin, K. Cytokines in Cancer Immunotherapy. Cancers 2011, 3, 3856–3893, doi:10.3390/cancers3043856.
- Peggs, K.S.; Quezada, S.A.; Allison, J.P. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin. Exp. Immunol. 2009, 157, 9–19, doi:10.1111/j.1365-2249.2009.03912.x.
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic Cancer Vaccines. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2013; Volume 119, pp. 421–475. ISBN 9780124071902.
- Bhatt, D.; Daemen, T. Therapeutic Vaccines and Cancer Immunotherapy. Vaccines 2020, 8, 596, doi:10.3390/vaccines8040596.
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B.A.G. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935, doi:10.1016/j.molonc.2015.10.018.
- Williams, A.D.; Payne, K.K.; Posey, A.D.; Hill, C.; Conejo-Garcia, J.; June, C.H.; Tchou, J. Immunotherapy for Breast Cancer: Current and Future Strategies. Curr. Surg. Rep. 2017, 5, 31, doi:10.1007/s40137-017-0194-1.
- Menon, S.; Shin, S.; Dy, G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers 2016, 8, 106, doi:10.3390/cancers8120106.
- Ti, D.; Bai, M.; Li, X.; Wei, J.; Chen, D.; Wu, Z.; Wang, Y.; Han, W. Adaptive T cell immunotherapy in cancer. Sci. China Life Sci. 2020, 468, doi:10.1007/s11427-020-1713-9.
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.-M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913, doi:10.1093/annonc/mdz398.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371, doi:10.3389/fimmu.2013.00371.
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009, doi:10.3390/ijms21145009.
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551, doi:10.1016/j.apsb.2018.03.001.
- Tang, H.; Qiao, J.; Fu, Y.-X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90, doi:10.1016/j.canlet.2015.10.009.
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, doi:10.3389/fimmu.2020.01109.
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63, doi:10.1016/j.coi.2015.01.011.
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218, doi:10.1038/s41573-018-0007-y.
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855, doi:10.1158/1078-0432.CCR-16-0049.
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 4, doi:10.1186/s40364-018-0116-0.
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. JNCI J. Natl. Cancer Inst. 2019, 111, 646–654, doi:10.1093/jnci/djz017.
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508, doi:10.1158/2326-6066.CIR-15-0231.
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283, doi:10.1038/s41467-020-16160-5.
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137, doi:10.1016/j.omto.2018.10.006.
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468, doi:10.1073/pnas.1524155113.
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458, doi:10.1073/pnas.1524193113.
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res. 2012, 18, 6436–6445, doi:10.1158/1078-0432.CCR-12-1449.
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852, doi:10.1158/0008-5472.CAN-11-3890.
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11, doi:10.1016/j.cell.2018.03.038.
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324, doi:10.1038/nchembio.2565.
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov. 2018, 8, 972–987, doi:10.1158/2159-8290.CD-17-1298.
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther. 2014, 22, 1211–1220, doi:10.1038/mt.2014.47.
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.P.; Cleary, S.; van der Stegen, S.J.C.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective Expansion of Chimeric Antigen Receptor-targeted T-cells with Potent Effector Function using Interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544, doi:10.1074/jbc.M110.127951.
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590, doi:10.1158/0008-5472.CAN-15-2524.
- Shin, J.H.; Park, H.B.; Oh, Y.M.; Lim, D.P.; Lee, J.E.; Seo, H.H.; Lee, S.J.; Eom, H.S.; Kim, I.-H.; Lee, S.H.; et al. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 2012, 119, 5678–5687, doi:10.1182/blood-2011-09-380519.
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243, doi:10.1186/s40425-019-0721-y.
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058, doi:10.1038/s41587-019-0192-1.
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75, doi:10.1038/nbt.2459.
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172, doi:10.1126/scitranslmed.3006597.
- Wilkie, S.; van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070, doi:10.1007/s10875-012-9689-9.
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419-432.e16, doi:10.1016/j.cell.2016.09.011.
- Ho, P.; Ede, C.; Chen, Y.Y. Modularly Constructed Synthetic Granzyme B Molecule Enables Interrogation of Intracellular Proteases for Targeted Cytotoxicity. ACS Synth. Biol. 2017, 6, 1484–1495, doi:10.1021/acssynbio.6b00392.
- Hamburger, A.E.; DiAndreth, B.; Cui, J.; Daris, M.E.; Munguia, M.L.; Deshmukh, K.; Mock, J.-Y.; Asuelime, G.E.; Lim, E.D.; Kreke, M.R.; et al. Engineered T cells directed at tumors with defined allelic loss. Mol. Immunol. 2020, doi:10.1016/j.molimm.2020.09.012.
- Abbas, A.K.; Trotta, E.; R. Simeonov, D.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482, doi:10.1126/sciimmunol.aat1482.
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042, doi:10.1126/science.aar3246.
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247, doi:10.1158/2159-8290.CD-17-0538.
- Kerkar, S.P.; Goldszmid, R.S.; Muranski, P.; Chinnasamy, D.; Yu, Z.; Reger, R.N.; Leonardi, A.J.; Morgan, R.A.; Wang, E.; Marincola, F.M.; et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Invest. 2011, 121, 4746–4757, doi:10.1172/JCI58814.
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A. V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-Infiltrating Lymphocytes Genetically Engineered with an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin. Cancer Res. 2015, 21, 2278–2288, doi:10.1158/1078-0432.CCR-14-2085.
- Zhang, L.; Davies, J.S.; Serna, C.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A.; Hinrichs, C.S. Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. J. Immunother. Cancer 2020, 8, e000210, doi:10.1136/jitc-2019-000210.
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2018, 7, e1378842, doi:10.1080/2162402X.2017.1378842.
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207, doi:10.4049/jimmunol.1800033.
- Hurton, L. V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797, doi:10.1073/pnas.1610544113.
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033, doi:10.1016/j.celrep.2017.09.002.
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459, doi:10.1038/s41587-019-0398-2.
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131, doi:10.1097/CCM.0000000000002053.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554, doi:10.1056/NEJMoa1708566.
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M. V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638, doi:10.1016/j.bbmt.2018.12.758.
- Maus, M. V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31, doi:10.1158/2326-6066.CIR-13-0006.
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254, doi:10.1182/blood-2004-11-4564.
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683, doi:10.1056/NEJMoa1106152.
- Griffioen, M.; van Egmond, E.H.M.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009, 94, 1316–1320, doi:10.3324/haematol.2008.001677.
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-Transduced T Lymphocytes as an Alternative Suicide Gene Therapy Approach for the Treatment of Graft-Versus-Host Disease. Hum. Gene Ther. 2004, 15, 63–76, doi:10.1089/10430340460732463.
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263, doi:10.1182/blood-2011-02-337360.
- Zhao, B.; Wang, Y.; Tan, X.; Zheng, X.; Wang, F.; Ke, K.; Zhang, C.; Liao, N.; Dang, Y.; Shi, Y.; et al. An Optogenetic Controllable T Cell System for Hepatocellular Carcinoma Immunotherapy. Theranostics 2019, 9, 1837–1850, doi:10.7150/thno.27051.
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2018, 6, 47–58, doi:10.1158/2326-6066.CIR-17-0126.
- Sakemura, R.; Terakura, S.; Watanabe, K.; Julamanee, J.; Takagi, E.; Miyao, K.; Koyama, D.; Goto, T.; Hanajiri, R.; Nishida, T.; et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol. Res. 2016, 4, 658–668, doi:10.1158/2326-6066.CIR-16-0043.
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833, doi:10.1038/srep39833.
- Allen, M.E.; Zhou, W.; Thangaraj, J.; Kyriakakis, P.; Wu, Y.; Huang, Z.; Ho, P.; Pan, Y.; Limsakul, P.; Xu, X.; et al. An AND-Gated Drug and Photoactivatable Cre- loxP System for Spatiotemporal Control in Cell-Based Therapeutics. ACS Synth. Biol. 2019, 8, 2359–2371, doi:10.1021/acssynbio.9b00175.
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.-J.; Wang, X.; Chien, S.; Wang, Y. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv. 2020, 6, eaay9209, doi:10.1126/sciadv.aay9209.

