 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peter Thompson | + 5214 word(s) | 5214 | 2021-01-05 09:49:42 | | | |
| 2 | Rita Xu | -2273 word(s) | 2941 | 2021-01-12 05:19:34 | | |
Video Upload Options
Type 1 diabetes (T1D) is a chronic metabolic disease characterized by insulin deficiency, generally resulting from progressive autoimmune-mediated destruction of pancreatic beta cells. While the phenomenon of beta cell autoimmunity continues to be an active area of investigation, recent evidence suggests that beta cell stress responses are also important contributors to disease onset.
1. Introduction
1.1. Autoimmune Type 1 Diabetes
Autoimmune type 1 diabetes (T1D) mellitus (also referred to as type 1A diabetes) results from insulin deficiency due to autoimmune-mediated destruction of pancreatic beta cells [1]. It often is distinguished from the less common type 1B diabetes, or idiopathic/nonautoimmune diabetes, in which the latter shows insulin deficiency and beta cell loss in the absence of beta cell autoimmunity [2]. T1D incidence has been increasing worldwide for the past few decades [3][4], and while it has a long-standing reputation as a pediatric disease, more recently an increasing number of young adults have been diagnosed [1][3]. Children and youth with T1D often experience challenges with insulin dosing to maintain optimal glycemic control as they age. Hence long-term diabetes-related complications such as nephropathy, neuropathy and retinopathy may be observed earlier in their lifetimes [1]. Additionally, despite modern improvements in insulins, which have increased the healthy lifespan of people living with T1D, there are growing financial barriers to affordable access in many countries [5][6] and an overall high risk of cardiovascular disease development, which is a major cause of death among older people living with T1D [7]. Currently there is no way to prevent or cure T1D, and daily insulin administration is the only safe and effective way to manage the disease. Thus, in addition to ongoing clinical care for those living with the disease, there is also an urgent need to better understand T1D pathogenesis and develop preventive therapies and treatments.
1.2. Stages in T1D Pathogenesis
Clinical heterogeneity in T1D patients is thought to arise as a result of different environmental exposures during development and genetic factors, each of which play a significant role in precipitating beta cell autoimmunity [3][8]. Models for the natural history of T1D have been extensively refined over the past decades as a consensus view has emerged [9][10][11]. In brief, three distinct clinical stages of disease progression have been recognized, although the timing and onset of each varies. During the earliest disease stage, patients are asymptomatic and as a result of environmental trigger(s) and genetic susceptibility, they develop beta cell autoimmunity (seroconversion) indicated by the presence of one or more autoantibodies against beta cell antigens, commonly insulin (INS), glutamate decarboxylase 65 (GAD65), islet antigen 2 (IA-2) and zinc transporter 8 (ZNT8) [12]. Remarkably, this early asymptomatic stage can precede a T1D diagnosis for years, and an increased number of autoantibodies correlates well with increased risk for T1D onset [13][14]. Newly developed risk scores incorporating genetic, epidemiological and immunological factors are greatly increasing predictive power for T1D onset in children between the ages of two to eight years old [15]. Stage 2 is characterized by declining beta cell function and/or mass as evidenced by abnormal glucose tolerance and sometimes very mild hyperglycemia [1]. However, overt hyperglycemia and the classical diabetes symptoms of polydipsia, polyuria and polyphagia are absent. Recent evidence suggests that beta cell dysfunction, rather than exclusively loss of beta cell mass during this period, may be a critical factor for disease progression [16]. Eventually, in the third stage, patients progress to become fully symptomatic, where functional beta cell mass is insufficient to meet normal metabolic demands leading to persistent hyperglycemia and the classical diabetes symptoms with or without diabetic ketoacidosis.
Interestingly, a honeymoon period has been described in an estimated 50% of new onset pediatric patients where their symptoms seem to improve and they experience clinical remission of diabetes upon the first administration of insulin and the subsequent reduction in dosage [17]. However, this phase is inevitably short-lived, usually lasting a few months, and patients subsequently require insulin again. This phenomenon is very poorly understood, but may suggest avenues for the proper timing of therapies to recover beta cell function in the long term after diagnosis [18]. Remarkably, although it was once believed that all beta cells are destroyed in T1D, recent work indicates that even in established T1D cases (> three years after diagnosis), proinsulin secretion persists for years in nearly all patients [19], and a substantial proportion of beta cells remain in many patients [20][21][22][23]. These observations hold promise for efforts to restore beta cell function well after diagnosis.
1.3. T1D as a Disease of the Immune System and Beta Cells
Historically, T1D has been viewed a disease of the immune system [24] where the beta cells are passive targets destroyed by a complex autoimmune process mediated by self-reactive cytotoxic CD4+ and CD8+ T cells supported by innate immunity. As a result of this emphasis, clinical interventions for the prevention and treatment of T1D have focused on immune-targeting therapies, some of which have shown beneficial effects [25][26]. For example, a recent clinical trial using a nondepleting anti-CD3 antibody (teplizumab), which targets T cells, in relatives of patients with T1D who were themselves at high risk of developing the disease (≥ 2 auto-antibodies and early signs of dysglycemia) led to a ~three year median delay in the progression to T1D onset when compared to a placebo [26]. However, the exact mechanisms of action of teplizumab are still unclear, and this therapeutic antibody also had little effect in some patients (e.g., non-responders) [26]. Similarly, a recent trial using golimumab, a therapeutic monoclonal tumor necrosis factor alpha (TNF-α) antibody, led to increased residual beta cell function and reduced insulin usage in new onset pediatric and young adult patients with T1D as compared with placebo [27]. This study also reported an increased number of hypoglycemic events along with increased frequency of infections in the golimumab patients [27]. Thus, while some immunotherapies can delay disease progression during stage two or even after onset in stage three, there are patients who do not respond and there are often unintended consequences of systemic immune modulation. A wide variety of immunotherapy clinical trials for new onset T1D or T1D prevention/delay are underway and include immune-modulating antibodies, cytokines, vaccines and regulatory T cell therapies [28].
Building from the classical paradigm of T1D as an autoimmune disease, a growing body of evidence supports the idea that beta cell dysfunction is just as critical as the autoimmune process, and that T1D is also a disease of the beta cells/islets [28][29]. Genome-wide association studies (GWAS) indicate that the majority of polymorphisms outside of the human leukocyte antigen (HLA) complex that are associated with T1D reside in genes known to be expressed in beta cells, including the INS gene itself [30]. Clinical observations over the past several years support the notion of ongoing beta cell dysfunction prior to diagnosis, and persistent beta cell mass and function, even in established T1D years after diagnosis [19][20][21][31][32][33]. Thus, an new emphasis on beta cell drug therapies could be an exciting avenue to reduce beta cell death, restore beta cell function and avert T1D onset during stage two or early into stage three of the disease [16].
2. Beta Cell Dysfunction in T1D
2.1. Beta Cell Endoplasmic Reticulum Stress, Unfolded Protein Response and Apoptosis
Perhaps the most well studied and widely regarded state of beta cell dysfunction during the pathogenesis of T1D is endoplasmic reticulum (ER) stress leading to apoptosis [34] (Figure 1A). Apoptosis is a form of programmed cell death triggered via a variety of mechanisms including internally as a result of irreparable cellular damage (intrinsic pathway), or externally as a result of surface receptor interactions with immune cells (extrinsic pathway) or as a result of the perforin-granzyme pathway [35] (for a detailed review of cell death mechanisms and nomenclature see [36]).
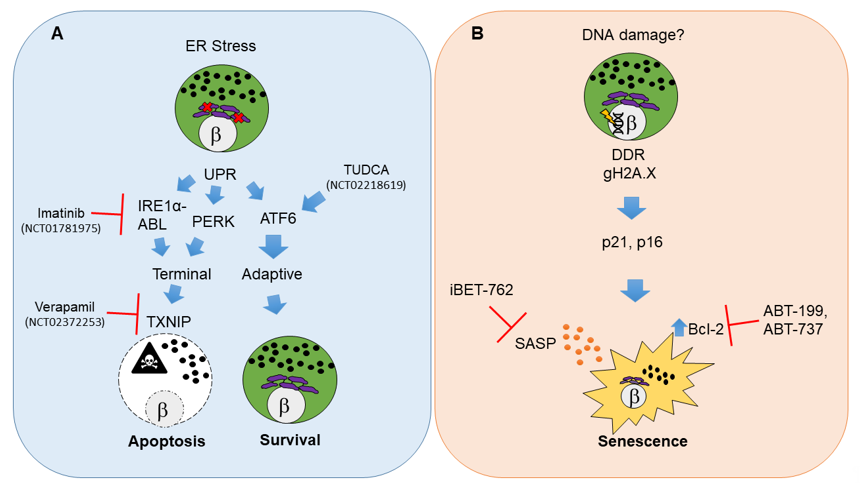
Figure 1. Molecular pathways and therapeutic targets for beta cell unfolded protein response (UPR)-mediated apoptosis and senescence in type 1 diabetes (T1D). (A) Beta cell apoptosis in T1D results from persistent endoplasmic reticulum (ER) stress that leads to activation of UPR master regulators IRE1α, PERK and ATF6. IRE1α mediates its functions through its RNAse and kinase activities that are potentiated by the Abelson tyrosine-protein kinase (ABLs). The balance of each UPR regulator dictates the outcome on beta cell fate. Unrelieved ER stress signals through IRE1α and PERK and shifts the pathway towards a terminal UPR and apoptosis mediated by thioredoxin interacting protein (TXNIP), whereas ATF6 is the major mediator of adaptive UPR leading to beta cell survival. Clinical trials in new onset adult T1D patients have used Verapamil, Imatinib or tauroursodeoxycholic acid (TUDCA) to attenuate terminal UPR and apoptosis and/or enhance adaptive UPR to delay the decline in residual beta cell function. (B) Beta cell senescence in T1D may be initiated by unresolved DNA damage (although the precise triggers of DNA damage remain unknown). A persistent DNA damage response (DDR) in beta cells is indicated by gH2A.X which is mediated by ATM. DNA damaged beta cells show activation of cyclin-dependent kinase inhibitors p21 and p16, which enforce a senescent growth arrest. Senescent beta cells upregulate the antiapoptotic protein Bcl-2 and develop a senescence-associated secretory phenotype (SASP). Small molecule inhibitors including senolytic compounds targeting Bcl-2 (ABT-199, ABT-737) or suppressing SASP at the level of gene expression (iBET-762) mitigate the deleterious effects of accumulated senescent beta cells in NOD mice and prevent T1D. These drugs have not been tested in clinical trials for T1D. The white circles and the β symbol indicate the nucleus, while the purple structure is the ER and black dots indicate insulin granules.
As beta cells have high demands for insulin synthesis, processing, folding and secretion, metabolic and immune-mediated stress are believed to directly impact the ability to sustain these processes [23]. As a consequence, a major cause of apoptosis in beta cells is ER stress-mediated activation of the unfolded protein response (UPR) [37]. Accordingly, decreased Ins1 gene dosage transiently improves beta cell ER function and relieves basal UPR stress in mice [38]. The UPR is a three-branched system that can either enable cells to maintain homeostasis (adaptive UPR) or lead them to commit to apoptosis (terminal UPR) [39]. Adaptive UPR signaling allows beta cells to cope with the stress of unfolded/misfolded proteins in the ER and recover, whereas a terminal UPR occurs if the stress is too great or prolonged, triggering apoptosis [40] (Figure 1A). Although recent work has focused on terminal UPR signaling as the major mechanism driving beta cell apoptosis, evidence from the widely studied nonobese diabetic (NOD) mouse model for human T1D [41] suggests that beta cells also undergo apoptosis via a combination of both the extrinsic pathway and perforin-granzyme pathway directed by cytotoxic T cells [42][43][44][45]. Similarly, a number of studies on human donor pancreas tissue have supported the idea that beta cells are destroyed in a heterogeneous fashion over the pancreas by CD8+ T cell-mediated cytotoxicity [20][21][33][46][47].
Another form of beta cell death suggested to be involved in T1D is necrosis [34], a lesser known form of cell death resulting from extensive damage, where cells are lysed and cellular contents are extruded to the extracellular environment, provoking immune activation and inflammatory responses [48]. This contrasts with what is thought to occur during apoptosis, as apoptotic cells are typically very short-lived and eliminated by phagocytes, leading to tissue remodeling [35][48]. While ongoing necrotic beta cell death is an attractive explanation for autoantigen release as has been proposed [49][50], the evidence for necrotic beta cell death as a putative mechanism in T1D is inconclusive.
A major open question in this area concerns which forms of beta cell death predominate in T1D, and whether beta cell death is generally continuous, relapsing-remitting or purely situational and context dependent [51]. Interestingly, a recent study using DNA methylation as a biomarker for circulating cell-free DNA (cfDNA) originating from beta cells found no evidence to substantiate ongoing beta cell death (where death was measured as beta cell-derived cfDNA in serum) in seroconverted individuals or those with recent onset or established T1D, whereas the same bioassay was sensitive enough to detect beta cell death following islet transplantation [52]. Thus, the various forms of beta cell death during the development of T1D, whatever they may be, either differ from those during islet transplantation or are simply not continuously occurring. As our knowledge of cell death mechanisms continues to expand [36], it will be important to elucidate additional pathways of beta cell death in T1D.
2.1.1. Pathways for UPR-Mediated Beta Cell Apoptosis
In beta cells, adaptive and terminal UPR are held in balance downstream of ER stress. ER stress activates the tripartite UPR pathway involving master regulators inositol requiring enzyme 1 alpha (IRE1α), PKR-like ER Kinase (PERK) and activating transcription factor 6 (ATF6), each of which regulate processes that control the apoptotic versus survival fate decision [37]. Notably, mRNA and protein markers of ER stress and UPR activation in beta cells are apparent in early-stage prediabetic NOD mice and human T1D donor pancreas sections [53][54][55]. When ER stress is prolonged or beyond remediation, there is a shift from adaptive to terminal UPR via IRE1α or PERK-dependent activation of the redox protein thioredoxin interacting protein (TXNIP) in beta cells [40][56] (Figure 1A). TXNIP activation is necessary to trigger the intrinsic apoptotic pathway in beta cells [40][56]. Accordingly, terminal UPR and apoptosis in beta cells can be averted with small molecule inhibitors targeting the RNAse activity of IRE1α or its binding partner, Abelson tyrosine-protein kinase (ABL) [57][58]. Recent genetic evidence indicates that IRE1α also controls beta cell identity, and beta cell-specific knockout of this UPR mediator protects against T1D in NOD mice [59]. Similarly, Txnip knockout prevents induction of apoptosis in rodent beta cell lines and islets ex vivo under conditions of persistent ER stress, such as high glucose [60].
2.1.2. Clinical Trials for UPR Therapies in T1D
Clinical trials testing beta cell-directed drug therapies for T1D (e.g., where beta cells are the primary target of the experimental agent, excluding transplantation) are few and far between. As of November 2020, on the clinicaltrials.gov website there were over 2,100 listed interventional T1D trials (inclusive of all trial statuses), yet only around 100 of these involve beta cells as drug targets, most of which are repurposing drugs currently used to treat type 2 diabetes (T2D). Interventional clinical trials utilizing small molecule drugs (alongside standard insulin regimens) to mitigate beta cell apoptosis in T1D in adults (≥ 18 years old) have shown promising early results in small cohorts. A recent phase II placebo-controlled clinical trial using daily Verapamil (TXNIP inhibitor) in recent onset T1D in adult patients over 12 months (NCT02372253) demonstrated enhanced preservation of beta cell function, reduced hypoglycemic events and decreased insulin requirements [61] (Figure 1A). However, the study size was quite limited (n = 11 participants for each treatment group) and also reported a high frequency of gastrointestinal side-effects and nausea, making it unclear whether this drug would be tolerable in a pediatric population. Similarly, a recent clinical trial using daily imantinib (IRE1α-ABL inhibitor) in recent onset T1D patients (NCT01781975) demonstrated a partial preservation of beta cell function compared with placebo over a one-year follow-up period (study has not been published) (Figure 1A). However, a broad range of adverse side-effects were reported and occurred more frequently in the imatinib-dosed group, categorized as gastrointestinal, skin, respiratory, cardiac, endocrine and infectious, suggesting a wide range of off-target effects. Indeed, it was recently reported that in addition to terminal UPR signaling, imatinib also directly affects insulin secretion from beta cells [62] and promotes reactive oxygen species (ROS) scavenging by B cells in NOD mice, an effect which is essential for diabetes reversal [63]. Thus, it would seem that there is still much to learn about the mechanisms of this drug.
Terminal UPR and apoptosis may also be averted by enhancing the ability of beta cells to handle unfolded proteins. The bile acid derivative tauroursodeoxycholic acid (TUDCA) acts as an ER stress inhibitor and protein chaperone [64] and prevents diabetes in the NOD mouse model in an ATF6-dependent manner [54] (Figure 1A). Notably, TUDCA-related acids have been safely used in infants and children for some time now as a treatment for various hepatobilliary diseases [65][66], suggesting they would also be safe for pediatric patients. A placebo-controlled phase II clinical trial for TUDCA in recent-onset adult T1D patients (NCT02218619) has recently completed, although the results await publication. The findings of this study will be important in providing further clinical evidence for the potential of UPR inhibitor therapies to enhance beta cell survival and function in T1D.
While these studies are indeed promising, a key feature of each of these drug therapies is that they require continuous dosing to inhibit their targets and be effective (daily dosing regimens were used in each of these trials). This kind of regimen often maximizes the extent and severity of side-effects. Indeed, uptake of these UPR inhibitory drugs in cell types other than ER-stressed beta cells would be detrimental when terminal UPR and apoptosis are required for tissue regeneration and cell turnover. Nevertheless, the evidence from these clinical trials suggests that even at diagnosis there is a clear window to improve, or at least delay, the decline of residual beta cell function beyond insulin therapy alone. The question of whether beta cell function can be improved in T1D by repurposing T2D drugs remains open. However recent studies targeting glucagon-like peptide 1 (GLP-1) and GLP-1 receptor (GLP1R) signaling suggest that this may not be effective (NCT01155284, NCT02284009) [67][68]. As future studies begin to understand the points at which beta cells are most vulnerable to ER stress-induced functional decline and terminal UPR during the various stages of T1D development, it may be possible to use these therapies intermittently and when they are most needed, obviating the side-effects resulting from chronic daily administration.
References
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Rev. Dis. Prim. 2017, 3, 17016, doi:10.1038/nrdp.2017.16.
- Catarino, D.; Silva, D.; Guiomar, J.; Ribeiro, C.; Ruas, L.; Cardoso, L.; Paiva, I. Non-immune-mediated versus immune-mediated type 1 diabetes: Diagnosis and long-term differences—Retrospective analysis. Metab. Syndr. 2020, 12, 1–6, doi:10.1186/s13098-020-00563-x.
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Rev. Endocrinol. 2019, 15, 635–650, doi:10.1038/s41574-019-0254-y.
- Dabelea, D.; Mayer-Davis, E.J.; Saydah, S.; Imperatore, G.; Linder, B.; Divers, J.; Bell, R.; Badaru, A.; Talton, J.W.; Crume, T.; et al. Prevalence of Type 1 and Type 2 Diabetes Among Children and Adolescents From 2001 to 2009. JAMA 2014, 311, 1778–1786, doi:10.1001/jama.2014.3201.
- Beran, D.; Mirza, Z.; Dong, J. Access to insulin: Applying the concept of security of supply to medicines. World Health Organ. 2019, 97, 358–364, doi:10.2471/blt.18.217612.
- The Lancet Diabetes & The bare essentials: Ensuring affordable access to insulin. Lancet Diabetes Endocrinol. 2017, 5, 151, doi:10.1016/s2213-8587(17)30038-4.
- Sharma, H.; Lencioni, M.; Narendran, P. Cardiovascular disease in type 1 diabetes. Endocrinol. Metab. 2019, 8, 28–34, doi:10.1097/xce.0000000000000167.
- Norris, J.M.; Johnson, R.K.; Stene, L.C. Type 1 diabetes—Early life origins and changing epidemiology. Lancet Diabetes Endocrinol. 2020, 8, 226–238, doi:10.1016/s2213-8587(19)30412-7.
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974, doi:10.2337/dc15-1419.
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300, doi:10.1038/nature08933.
- Eisenbarth, G.S. Type I Diabetes Mellitus. Engl. J. Med. 1986, 314, 1360–1368, doi:10.1056/nejm198605223142106.
- Lampasona, V.; Liberati, D. Islet Autoantibodies. Diabetes Rep. 2016, 16, 53, doi:10.1007/s11892-016-0738-2.
- Regnell, S.E.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381, doi:10.1007/s00125-017-4308-1.
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA 2013, 309, 2473–2479, doi:10.1001/jama.2013.6285.
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, Å.; Rewers, M.J.; She, J.X.; Ziegler, A.G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A combined risk score enhances prediction of type 1 diabetes among susceptible children. Med. 2020, 26, 1247–1255, doi:10.1038/s41591-020-0930-4.
- Sims, E.K.; Mirmira, R.G.; Evans-Molina, C. The role of beta-cell dysfunction in early type 1 diabetes. Opin. Endocrinol. Diabetes Obes. 2020, 27, 215–224, doi:10.1097/med.0000000000000548.
- Nwosu, B.U. Partial Clinical Remission of Type 1 Diabetes Mellitus in Children: Clinical Applications and Challenges with its Definitions. Med. J. Diabetes 2019, 4, 89–98.
- Zhong, T.; Tang, R.; Gong, S.; Li, J.; Li, X.; Zhou, Z. The remission phase in type 1 diabetes: Changing epidemiology, definitions, and emerging immuno‐metabolic mechanisms. Diabetes/Metab. Res. Rev. 2020, 36, 1–7, doi:10.1002/dmrr.3207.
- Sims, E.K.; Bahnson, H.T.; Nyalwidhe, J.; Haataja, L.; Davis, A.K.; Speake, C.; DiMeglio, L.A.; Blum, J.; Morris, M.A.; Mirmira, R.G.; et al. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care 2019, 42, 258–264, doi:10.2337/dc17-2625.
- Wang, Y.J.; Traum, D.; Schug, J.; Gao, L.; Liu, C.; Atkinson, M.A.; Powers, A.C.; Feldman, M.D.; Naji, A.; Chang, K.M.; et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019, 29, 769–783.e4, doi:10.1016/j.cmet.2019.01.003.
- Damond, N.; Engler, S.; Zanotelli, V.R.T.; Schapiro, D.; Wasserfall, C.H.; Kusmartseva, I.; Nick, H.S.; Thorel, F.; Herrera, P.L.; Atkinson, M.A.; et al. A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab. 2019, 29, 755–768.e5, doi:10.1016/j.cmet.2018.11.014.
- Lam, C.J.; Jacobson, D.R.; Rankin, M.M.; Cox, A.R.; Kushner, J.A. β Cells Persist in T1D Pancreata Without Evidence of Ongoing β-Cell Turnover or Neogenesis. Clin. Endocrinol. Metab. 2017, 102, 2647–2659, doi:10.1210/jc.2016-3806.
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Rev. Endocrinol. 2020, 16, 349–362, doi:10.1038/s41574-020-0355-7.
- Gale, E.A.M. The discovery of type 1 diabetes. Diabetes 2001, 50, 217–226, doi:10.2337/diabetes.50.2.217.
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (Anti-CD3 mAb) Treatment Preserves C-Peptide Responses in Patients with New-Onset Type 1 Diabetes in a Randomized Controlled Trial: Metabolic and Immunologic Features at Baseline Identify a Subgroup of Responders. Diabetes 2013, 62, 3766–3774, doi:10.2337/db13-0345.
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. Engl. J. Med. 2019, 381, 603–613, doi:10.1056/nejmoa1902226.
- Quattrin, T.; Haller, M.J.; Steck, A.K.; Felner, E.I.; Li, Y.; Xia, Y.; Leu, J.H.; Zoka, R.; Hedrick, J.A.; Rigby, M.R.; et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. Engl. J. Med. 2020, 383, 2007–2017, doi:10.1056/nejmoa2006136.
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64, doi:10.1016/s2213-8587(18)30112-8.
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Rev. Endocrinol. 2020, doi:10.1038/s41574-020-00443-4.
- Pociot, F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Transl. Immunol. 2017, 6, e162, doi:10.1038/cti.2017.51.
- Evans-Molina, C.; Sims, E.K.; DiMeglio, L.A.; Ismail, H.M.; Steck, A.K.; Palmer, J.P.; Krischer, J.P.; Geyer, S.; Xu, P.; Sosenko, J.M. β Cell dysfunction exists for more than 5 years prior to type 1 diabetes diagnosis. JCI Insight 2018, 3, e120877, doi:10.1172/jci.insight.120877.
- Oram, R.A.; McDonald, T.J.; Shields, B.M.; Hudson, M.M.; Shepherd, M.H.; Hammersley, S.; Pearson, E.R.; Hattersley, A.T.; Sanders, T.; Tiley, S.; et al. Most People With Long-Duration Type 1 Diabetes in a Large Population-Based Study Are Insulin Microsecretors. Diabetes Care 2015, 38, 323–328, doi:10.2337/dc14-0871.
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731, doi:10.2337/db15-0779.
- Wilcox, N.S.; Rui, J.; Hebrok, M.; Herold, K.C. Life and death of Beta cells in Type 1 diabetes: A comprehensive review. Autoimmun. 2016, 71, 51–58, doi:10.1016/j.jaut.2016.02.001.
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Pathol. 2007, 35, 495–516, doi:10.1080/01926230701320337.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541, doi:10.1038/s41418-017-0012-4.
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Metab. 2019, 27, S60–S68, doi:10.1016/j.molmet.2019.06.012.
- Szabat, M.; Page, M.M.; Panzhinskiy, E.; Skovsø, S.; Mojibian, M.; Fernandez-Tajes, J.; Bruin, J.E.; Bround, M.J.; Lee, J.T.C.; Xu, E.E.; et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces β Cell Proliferation. Cell Metab. 2016, 23, 179–193, doi:10.1016/j.cmet.2015.10.016.
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555, doi:10.1016/j.tcb.2013.06.005.
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264, doi:10.1016/j.cmet.2012.07.007.
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. Autoimmun. 2016, 66, 76–88, doi:10.1016/j.jaut.2015.08.019.
- Amrani, A.; Verdaguer, J.; Anderson, B.; Utsugi, T.; Bou, S.; Santamaria, P. Perforin-independent β-cell destruction by diabetogenic CD8+ T lymphocytes in transgenic nonobese diabetic mice. Clin. Investig. 1999, 103, 1201–1209, doi:10.1172/jci6266.
- Kägi, B.D.; Odermatt, B.; Seiler, P.; Zinkernagel, R.M.; Mak, T.W.; Hengartner, H. Perforin-deficient Nonobese Diabetic Mice. Exp. Med. 1997, 186, 989–997.
- Mohamood, A.S.; Guler, M.L.; Xiao, Z.; Zheng, D.; Hess, A.; Wang, Y.; Yagita, H.; Schneck, J.P.; Hamad, A.R.A. Protection from Autoimmune Diabetes and T-Cell Lymphoproliferation Induced by FasL Mutation Are Differentially Regulated and Can Be Uncoupled Pharmacologically. J. Pathol. 2007, 171, 97–106, doi:10.2353/ajpath.2007.070148.
- Thomas, H.E.; Kay, T.W. Intracellular pathways of pancreatic β-cell apoptosis in type 1 diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 790–796, doi:10.1002/dmrr.1253.
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; Von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. Exp. Med. 2012, 209, 51–60, doi:10.1084/jem.20111187.
- Panzer, J.K.; Hiller, H.; Cohrs, C.M.; Almaça, J.; Enos, S.J.; Beery, M.; Cechin, S.; Drotar, D.M.; Weitz, J.R.; Santini, J.; et al. Pancreas tissue slices from organ donors enable in situ analysis of type 1 diabetes pathogenesis. JCI Insight 2020, 5, doi:10.1172/jci.insight.134525.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592, doi:10.1002/cbin.11137.
- Graham, K.L.; Sutherland, R.M.; Mannering, S.I.; Zhao, Y.; Chee, J.; Krishnamurthy, B.; Thomas, H.E.; Lew, A.M.; Kay, T.W.H. Pathogenic Mechanisms in Type 1 Diabetes: The Islet is Both Target and Driver of Disease. Diabet. Stud. 2012, 9, 148–168, doi:10.1900/RDS.2012.9.148.
- Boldison, J.; Wong, F.S. Immune and Pancreatic β Cell Interactions in Type 1 Diabetes. Trends Endocrinol. Metab. 2016, 27, 856–867, doi:10.1016/j.tem.2016.08.007.
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82, doi:10.1016/s0140-6736(13)60591-7.
- Neiman, D.; Gillis, D.; Piyanzin, S.; Cohen, D.; Fridlich, O.; Moss, J.; Zick, A.; Oron, T.; Sundberg, F.; Forsander, G.; et al. Multiplexing DNA methylation markers to detect circulating cell-free DNA derived from human pancreatic β cells. JCI Insight 2020, 5, doi:10.1172/jci.insight.136579.
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet Beta-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes 2012, 61, 818–827, doi:10.2337/db11-1293.
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Transl. Med. 2013, 5, 211ra156.
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420, doi:10.1007/s00125-012-2604-3.
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-Interacting Protein Mediates ER Stress-Induced β Cell Death through Initiation of the Inflammasome. Cell Metab. 2012, 16, 265–273, doi:10.1016/j.cmet.2012.07.005.
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab. 2017, 25, 883–897.e8, doi:10.1016/j.cmet.2017.03.018.
- Louvet, C.; Szot, G.L.; Lang, J.; Lee, M.R.; Martinier, N.; Bollag, G.; Zhu, S.; Weiss, A.; Bluestone, J.A. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Natl. Acad. Sci. USA 2008, 105, 18895–18900, doi:10.1073/pnas.0810246105.
- Lee, H.; Lee, Y.S.; Harenda, Q.; Pietrzak, S.; Oktay, H.Z.; Schreiber, S.; Liao, Y.; Sonthalia, S.; Ciecko, A.E.; Chen, Y.G.; et al. Beta Cell Dedifferentiation Induced by IRE1α Deletion Prevents Type 1 Diabetes. Cell Metab. 2020, 31, 822–836.e5, doi:10.1016/j.cmet.2020.03.002.
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP Protects Against Mitochondria-Mediated Apoptosis but Not Against Fatty Acid-Induced ER Stress-Mediated β-Cell Death. Diabetes 2010, 59, 440–447, doi:10.2337/db09-0949.
- Ovalle, F.; Grimes, T.; Xu, G.; Patel, A.J.; Grayson, T.B.; Thielen, L.A.; Li, P.; Shalev, A. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Med. 2018, 24, 1108–1112, doi:10.1038/s41591-018-0089-4.
- Xia, C.Q.; Zhang, P.; Li, S.; Yuan, L.; Xia, T.; Xie, C.; Clare-Salzler, M.J. C-Abl Inhibitor Imatinib Enhances Insulin Production by β Cells: C-Abl Negatively Regulates Insulin Production via Interfering with the Expression of NKx2.2 and GLUT-2. PLoS ONE 2014, 9, e97694, doi:10.1371/journal.pone.0097694.
- Wilson, C.S.; Spaeth, J.M.; Karp, J.; Stocks, B.T.; Hoopes, E.M.; Stein, R.W.; Moore, D.J. B lymphocytes protect islet β cells in diabetes-prone NOD mice treated with imatinib. JCI Insight 2019, 4, doi:10.1172/jci.insight.125317.
- Kusaczuk, M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471, doi:10.3390/cells8121471.
- Lebensztejn, D.M. Application of ursodeoxycholic acid (UDCA) in the therapy of liver and biliary duct diseases in children. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2000, 6, 632–636.
- Heubi, J.E.; Wiechmann, D.A.; Creutzinger, V.; Setchell, K.D.R.; Squires, R.J.; Couser, R.; Rhodes, P. Tauroursodeoxycholic acid (TUDCA) in the prevention of total parenteral nutrition-associated liver disease. Pediatr. 2002, 141, 237–242, doi:10.1067/mpd.2002.125802.
- Pozzilli, P.; Bosi, E.; Cirkel, D.; Harris, J.; Leech, N.; Tinahones, F.J.; Vantyghem, M.C.; Vlasakakis, G.; Ziegler, A.G.; Janmohamed, S. Randomized 52-week Phase 2 Trial of Albiglutide Versus Placebo in Adult Patients With Newly Diagnosed Type 1 Diabetes. Clin. Endocrinol. Metab. 2020, 105, dgaa149, doi:10.1210/clinem/dgaa149.
- Griffin, K.J.; Thompson, P.A.; Gottschalk, M.; Kyllo, J.H.; Rabinovitch, A. Combination therapy with sitagliptin and lansoprazole in patients with recent-onset type 1 diabetes (REPAIR-T1D): 12-month results of a multicentre, randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2014, 2, 710–718, doi:10.1016/s2213-8587(14)70115-9.

