 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fatima Domenica Elisa De Palma | + 2144 word(s) | 2144 | 2021-01-07 11:33:58 | | | |
| 2 | Catherine Yang | -2 word(s) | 2142 | 2021-01-12 04:56:48 | | |
Video Upload Options
Cystic fibrosis (CF) is a lifelong disorder affecting 1 in 3500 live births worldwide. It is a monogenetic autosomal recessive disease caused by loss-of-function mutations in the gene encoding the chloride channel cystic fibrosis transmembrane conductance regulator (CFTR), the impairment of which leads to ionic disequilibria in exocrine organs.
1. Introduction
The dual role of time, as a fearsome enemy or important ally in the course of human diseases, has motivated researchers to advance in the frantic (and necessary) understanding of their cellular and molecular bases, hoping to identify novel approaches for patient care. The discovery of a variety of disease-modulatory molecules, including non-coding RNAs, that are present not only at the tissue level, but also in the bloodstream, has revolutionized the world of clinical molecular biology.
microRNAs (miRNAs), a class of small non-coding RNAs, are well known to regulate a diverse array of biological processes, such as proliferation, development, and metabolism [1][2]. They act as repressors of one or several genes by binding to complementary sites within the 3’ untranslated region (UTR) of target mRNAs [1][3][4].
miRNAs positively or negatively impact on the pathogenesis and/or progression of human diseases. They are (de)regulated by different mechanisms, such as epigenetic alterations, chromosomal abnormalities, as well as changes in transcriptional control [2][5]. Moreover, the altered abundance of miRNAs, along with their high stability (due to their small length of about 18–22 nucleotides) renders them highly potential biomarker candidates in non-invasive sources (i.e., blood, serum, urine, and saliva) [6][7].
The clinical potential of miRNAs as diagnostic and prognostic, as well as predictive biomarkers, has been widely described, especially in the context of human cancer [6][8][9][10]. Moreover, potential therapeutics harness the deregulation of miRNAs in disease conditions by means of miRNA mimics, so-called antimiRs [11][12].
Accumulating evidence suggests the involvement of miRNAs also in genetic diseases, including cystic fibrosis (CF) [13][14][15][16][17][18][19]. In particular, many studies showed the direct or indirect regulatory impact of deregulated miRNAs on the expression of the cystic fibrosis transmembrane conductance regulator (CFTR) mRNA [20]. Moreover, their participation in the control of the inflammation of CF airways has been demonstrated, along with their potential role as circulating biomarkers [17]. Thus, the contribution of miRNAs to disease progression and severity opens new and interesting scenarios for the clinical management of CF patients.
2. Cystic Fibrosis
2.1. Cystic Fibrosis, A Multisystemic Deadly Disease
CF, also known as mucoviscidosis, is one of the most common Mendelian autosomal recessive genetic disorders [21][22]. Its incidence varies according to ethnicity, affecting approximately 1 in about 2500 newborns in Europe [23].
CF is caused by mutations in CFTR, which belongs to the ATP binding cassette (ABC) gene family [22][24]. Environmental factors, as well as non-CFTR gene modifiers, influence the manifestations and progression of the disease [25].
The CFTR gene is located on the long arm of the chromosome 7 [25]. The CFTR protein is mostly located at the apical membranes of a variety of secretory ephitelia (which produce mucus, sweat, and digestive juices) harbored in multiple organs, including sweat glands, lungs, and pancreas, as well as the gut [21][25]. CFTR mainly functions as an anion channel; it acts as a cyclic adenosine monophosphate (cAMP)-dependent chloride channel and as bicarbonate channel. Moreover, the involvement of CFTR in the modulation of other ion channels, such as the epithelial sodium channel (ENaC), has been also described, not without controversy [26]. CFTR transports chloride and bicarbonate across secretory epithelia, thus regulating both the secretion and the absorption of salt and water and maintaining epithelial surface hydration.
Mutations in CFTR lead to the impairment of the expression, as well as of the function and stability at the mRNA and CFTR protein levels [27], thus unbalancing fluid and electrolyte homeostasis. For instance, an abnormal excessive secretion of salt from the sweat glands that has not been reabsorbed by the sweat duct cells is one of the main symptoms of CF [25]. At present, the measurement of chloride excreted in sweat is the gold standard to diagnose CF.
Another deleterious effect, which is due to the reduction of the bicarbonate release, is the formation of thickened and viscous secretions in bronchi, pancreas, biliary tract, intestine, and the reproductive system. Although mucus is an alley of innate immunity due to its composition made, inter alia, of antibacterial defensins and immunoglobulins, its hyperviscous form is pathogenic in CF [28].
Concerning the respiratory tract of CF patients, defects in fluid secretion and the reduced pH of the airway surface liquid affect ciliary beating and compromise mucociliary clearance, thus increasing the viscosity of the mucus but decreasing the activity of antimicrobial molecules [29]. As a consequence, mucus accumulates and obstructs the airways (bronchiectasis), promoting chronic bacterial infections and inflammatory lung damage. The airway microbiome changes across the ages [30]. Typically, CF adults are persistently colonized by Pseudomonas aeruginosa (P. aeruginosa) [31]. Altogether, these changes trigger acute pulmonary exacerbations and respiratory failure.
The gastrointestinal tract as well as the biliary ducts are also impacted by unbalanced ion secretion [32][33]. The more acidic intestinal environment, along with the sticky and thickened mucus, which is hardly cleared, alters the activity of digestive enzymes; this impairs the assimilation of digestive products, delays the intestinal transit, and causes bowel obstructions [32]. Meconium ileus is one of the earliest clinical manifestations and the most serious acute complication of CF at the intestinal level [34]. Nonetheless, older CF patients may also present distal intestinal obstruction syndrome (DIOS) and intussusception [33]. Furthermore, intestinal inflammation and dysbiosis occur in the context of CF [32][35].
Severe inflammation, viscous secretions, and fibrosis lead to pancreatic insufficiency, leading to abdominal pain, malabsorption, and weight loss [36]. Additionally, diabetes mellitus and male infertility may become manifest [37][38][39]. Congenital aplasia of vas deferens (CAVD) contributes to male infertility and obstructive azoospermia [40][41]. This abnormality of the urogenital tract has different clinical presentations, according, inter alia, to its bilaterally (CBAVD) or unilaterally (CUAVD) occurrence [40][41][42]. In women with CF, fertility is compromised due to thicker cervical secretions [37].
Although CF is a chronic multisystemic disease that is ultimately lethal, daily care and personalized (expensive) treatments extend the life expectancy of most CF patients until 40 years of age [43]. The medical care for CF requires multiple interventions including high-calorie ingestion, therapy based on the replacement of pancreatic enzymes and, most importantly, the management of the pulmonary exacerbations [21][25][44]. For instance, airway-clearance techniques and aerosolized mucolytic agents, along with administration of some specific antibiotics and anti-inflammatory drugs can avoid, reduce, or eradicate bacterial infections and improve lung functions [25].
In addition, targeted therapies for CF patients with specific CFTR pathogenic mutations have been developed [43][44][45][46]. These treatments are based on a class of compounds, the CFTR modulators, which are able to modulate (enhance or restore) the expression, function, and stability of mutant CFTR proteins [43][45]. Generally, CFTR modulators are grouped into five classes: (i) “potentiators”, which restore the channel gating and conductance of the mutant CFTR protein; (ii) “correctors”, which are drugs that rescue the protein folding, thus augmenting the traffic to the plasma surface of CFTR mutant; (iii) “stabilizers” that avoid the removal and degradation by lysosomes of CFTR through its stronger stabilization on the plasma membrane; (iv) “amplifiers” that increase the expression of CFTR at the mRNA level, and, in turn, the amount of CFTR protein; and (v) “read-through” agents that restore the functional full-length protein affected by premature termination codons. To date, only two classes of modulators, potentiators and correctors, have been approved to treat CF patients [45].
Finally, lung transplantation is reserved for end-stage CF patients, but it extends survival by only 5 years on average [47].
2.2. The Mutational Landscape of the Cystic Fibrosis
Since the discovery of the first CFTR mutation in 1989, to date, more than 2000 different genetic variants have been identified (www.CFTR2.org). However, only a few of them (about 350 mutations) are considered as pathogenic (www.CFTR2.org) [43].
Severe mutations belonging to classes I, II, and III [27] lead to the loss of the CFTR function. In particular, class I mutations abolish the synthesis of the protein. They include nonsense mutations (i.e., premature termination codons). Class II mutations cause the retention of a misfolded protein at the endoplasmic reticulum and its subsequent degradation by the proteasome. The most common mutation in Europe, the F508del, which consists in the deletion of a phenylalanine at position 508, belongs to this class. Class III mutations affect the regulation and gating of the CFTR channel.
By contrast, class IV to VI mutations confer a milder CF phenotype [27]. Class IV mutations decrease the conductance of chloride and bicarbonate ions. Class V mutations lead to a reduction of the abundance (synthesis or maturation) of the normal CFTR protein. Class VI mutations destabilize the protein at the cell surface by increasing the endocytosis and lysosomal degradation of CFTR.
A new categorization of CFTR mutations into VII classes according to the therapeutic strategies has been suggested by De Boeck and Amaral [46]. The only difference compared to the traditional classification system consists in the addition of class VII mutations, which have the same functional characteristics as class I mutations but cannot be rescued by pharmacological interventions [46].
3. microRNAs as Regulators of Inflammation in Cystic Fibrosis
Inflammation plays a critical role in the progression of CF [48]. As described before, the overproduction of hyperviscous mucus obstructs airways and impairs mucociliary clearance. This creates a perfect nutrient-rich environment for bacterial colonization as well as for pro-inflammatory modulators that lead to the progressive structural damage of the lung (Figure 1).
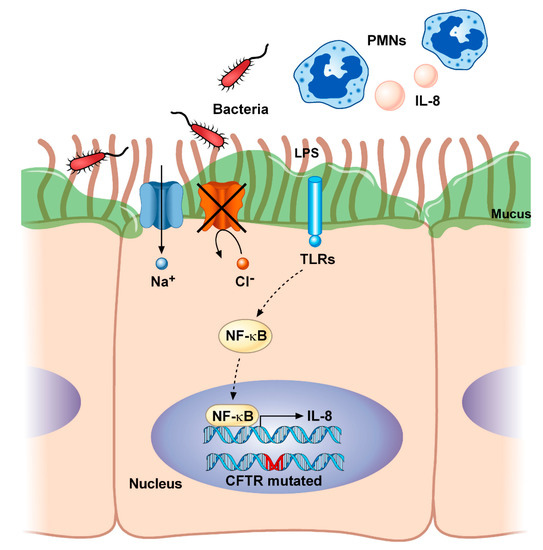
Figure 1. Airway inflammation in cystic fibrosis. Mutations in CFTR lead to imbalances in fluid and electrolyte homeostasis characterized by the lack of transport of chloride (Cl−) and excessive sodium (Na+) reabsorption. This impairs mucociliary clearance and increases the viscosity of the mucus, thus promoting chronic bacterial colonization (i.e., by Staphylococcus aureus and Pseudomonas aeruginosa). The resultant shedding of microbial molecules, known as pathogen-associated molecular patterns (PAMPs) (i.e., lipopolysaccharide, LPS), being recognized by the Toll-like receptors (TLRs), constitutively activates the NF-κB and causes the consequent production of inflammatory cytokines (i.e., IL-8), as well as the accumulation of polymorphonuclear neutrophils (PMNs) into the airways. CFTR, cystic fibrosis transmembrane conductance regulator; IL-8, interleukin-8; LPS, lipopolysaccharide; NF-κB, nuclear factor κ light-chain enhancer of activated B cells; PMNs, polymorphonuclear neutrophils; TLRs, toll-like receptors.
miRNAs can also control other signaling pathways to trigger inflammation in CF. High levels of miR-199a-5p were detected in human and murine CF macrophages and murine CF lungs [49]. The aberrant expression of miR-199a-5p, mediated by PI3K/AKT signaling, reduced the expression of caveolin 1 (CAV1), which is a mediator of inflammation processes, and in turn increased Toll-like receptor (TLR) 4. Moreover, the downregulation of miR-199a-5p was able to reduce the inflammation in CF macrophages via restoring the expression of CAV1. Furthermore, in vitro and in vivo studies demonstrated that the administration of the non-steroidal anti-inflammatory drug celecoxib rescued the altered pathway mediated by AKT/miR-199a-5p/CAV1 in CF macrophages and reduced lung inflammation in CFTR-deficient mice, respectively [49].
An elevated expression of miR-145, found in nasal airway cells from CF patients when compared to non-CF controls, correlated with the downregulation of SMAD3 [50]. SMAD3 is a negative modulator of the NF-kB–IL-8 pathway mediated by TGF-β1; therefore, miR-145 may contribute to CF-related inflammation [50].
Differently from the aforementioned miRNAs, miR-126 has an anti-inflammatory role in CF. It has been the first miRNA identified as deregulated in CF in 2010 [51]. In particular, low levels of miR-126 were found in CF airway epithelial cells. This downregulation correlated with the upregulation of the target of Myb protein 1 (TOM1), which is a negative regulator of TLR2, TLR4, IL-1, IL-1β, TNF-α, and NF-kB. When CF cells were stimulated with LPS and IL-1β, the knockdown of TOM1 significantly increased the NF-kB-mediated IL-8 secretion [51]. In addition, another study reported that miR-1343 attenuated the pathway of fibrosis by decreasing levels of activated TGF-β effector molecules, including phosphorylated (p) SMAD3 (pSMAD3) as well as pSMAD2 and consequently disturbing the cell migration and epithelial–mesenchymal transition [52].
Some miRNAs may also participate in chronic inflammation by affecting the remodeling of the pulmonary epithelium or by impacting the expression of genes involved in mucus hypersecretion. The role of miR-146a in CF inflammation has been linked to its negative impact on the production of the mucin 5AC (MUC5AC), which is a major component of the airway mucus [53]. Moreover, the knockdown of miR-146a in human bronchial epithelial cells resulted in the activation of the NF-kB and Jun N-terminal kinase (JNK) pathways [53].
In bronchial epithelial cells, miR-145, miR-494, and particularly miR-221 regulate the transcription factor 6 (ATF6), which is implicated in the airway inflammation through endoplasmic reticulum stress [54][55].
The deregulation of miR-31 enhanced the production of the cathepsin S (CTSS) via the direct inhibition of the interferon regulatory factor 1 (IRF-1) in CF epithelial cells [56]. CTSS is an elastinolytic and collagenolytic cysteine protease. High levels of CTSS in CF patients increased pulmonary neutrophilic infiltration of the lung, as well as the inactivation of antimicrobial proteins, such as lactoferrin and members of the β-defensin family, therefore contributing to lung inflammation and infection, and determining lung damage [57].
References
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433.
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9.
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94.
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114.
- Han, L.; Witmer, P.D.; Casey, E.; Valle, D.; Sukumar, S. DNA methylation regulates MicroRNA expression. Cancer Biol. 2007, 6, 1284–1288.
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469.
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276.
- Lan, H.; Lu, H.; Wang, X.; Jin, H. MicroRNAs as Potential Biomarkers in Cancer: Opportunities and Challenges. Available online: https://www.hindawi.com/journals/bmri/2015/125094/ (accessed on 3 September 2020).
- De Palma, F.D.E.; Luglio, G.; Tropeano, F.P.; Pagano, G.; D’Armiento, M.; Kroemer, G.; Maiuri, M.C.; De Palma, G.D. The Role of Micro-RNAs and Circulating Tumor Markers as Predictors of Response to Neoadjuvant Therapy in Locally Advanced Rectal Cancer. Int. J. Mol. Sci. 2020, 21, 7040.
- De Palma, F.D.E.; D’Argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers 2019, 11, 1017.
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222.
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638.
- Finotti, A.; Fabbri, E.; Lampronti, I.; Gasparello, J.; Borgatti, M.; Gambari, R. MicroRNAs and Long Non-coding RNAs in Genetic Diseases. Mol. Diagn. 2019, 23, 155–171.
- Narożna, B.; Langwiński, W.; Szczepankiewicz, A. Non-Coding RNAs in Pediatric Airway Diseases. Genes 2017, 8, 348.
- Greene, C.M. MicroRNA Expression in Cystic Fibrosis Airway Epithelium. Biomolecules 2013, 3, 157–167.
- Varilh, J.; Bonini, J.; Taulan-Cadars, M. Role of Non-coding RNAs in Cystic Fibrosis. Cyst. Fibros. Light New Res. 2015.
- Sonneville, F.; Ruffin, M.; Guillot, L.; Rousselet, N.; Le Rouzic, P.; Corvol, H.; Tabary, O. New insights about miRNAs in cystic fibrosis. Am. J. Pathol. 2015, 185, 897–908.
- Noel, S.; Leal, T. Emerging Roles of microRNAs in Cystic Fibrosis—From Pathogenesis to Development of New Therapies. Cyst. Fibros. Light New Res. 2015.
- Mitash, N.; Donovan, J.E.; Swiatecka-Urban, A. The Role of MicroRNA in the Airway Surface Liquid Homeostasis. Int. J. Mol. Sci. 2020, 21, 3848.
- McKiernan, P.J.; Greene, C.M. MicroRNA Dysregulation in Cystic Fibrosis. Available online: https://www.hindawi.com/journals/mi/2015/529642/ (accessed on 25 November 2020).
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531.
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56.
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589.
- Hwang, T.-C.; Kirk, K.L. The CFTR Ion Channel: Gating, Regulation, and Anion Permeation. Cold Spring Harb. Perspect. Med. 2013, 3.
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 1–19.
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115.
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38.
- Puchelle, E.; Bajolet, O.; Abély, M. Airway mucus in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 115–119.
- Chioccioli, M.; Feriani, L.; Kotar, J.; Bratcher, P.E.; Cicuta, P. Phenotyping ciliary dynamics and coordination in response to CFTR-modulators in Cystic Fibrosis respiratory epithelial cells. Nat. Commun. 2019, 10, 1763.
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes 2020, 11, 536.
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241.
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 175–185.
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753.
- Bagolan, P.; Morini, F.; Conforti, A. Meconium Ileus. In Pediatric Surgery: General Principles and Newborn Surgery; Puri, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 973–992. ISBN 978-3-662-43588-5.
- Hayden, H.S.; Eng, A.; Pope, C.E.; Brittnacher, M.J.; Vo, A.T.; Weiss, E.J.; Hager, K.R.; Martin, B.D.; Leung, D.H.; Heltshe, S.L.; et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat. Med. 2020, 26, 215–221.
- Madácsy, T.; Pallagi, P.; Maleth, J. Cystic Fibrosis of the Pancreas: The Role of CFTR Channel in the Regulation of Intracellular Ca2+ Signaling and Mitochondrial Function in the Exocrine Pancreas. Front. Physiol. 2018, 9.
- Casciaro, R.; Cresta, F.; Favilli, F.; Minicucci, L. Cystic Fibrosis and Fertility. Cyst. Fibros. Light New Res. 2015.
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 2018, 9.
- Wilson, C. Cystic fibrosis-related diabetes | Nature Reviews Endocrinology. Nat. Rev. Endocrinol. 2011, 7, 375.
- Bieth, E.; Hamdi, S.M.; Mieusset, R. Genetics of the congenital absence of the vas deferens. Hum. Genet. 2020.
- de Souza, D.A.S.; Faucz, F.R.; Pereira-Ferrari, L.; Sotomaior, V.S.; Raskin, S. Congenital Bilateral Absence of the Vas Deferens as an Atypical Form of Cystic Fibrosis: Reproductive Implications and Genetic Counseling. Andrology 2018, 6, 127–135.
- Wagenknecht, L.V.; Lotzin, C.F.; Sommer, H.-J.; Schirren, C. Vas Deferens Aplasia: Clinical and Anatomical Features of 90 Cases. Andrologia 1983, 15, 605–613.
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharm. 2020, 10.
- Maiuri, L.; Raia, V.; Kroemer, G. Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 2017, 24, 1825–1844.
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34.
- Boeck, K.D.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674.
- Ramos, K.J.; Smith, P.J.; McKone, E.F.; Pilewski, J.M.; Lucy, A.; Hempstead, S.E.; Tallarico, E.; Faro, A.; Rosenbluth, D.B.; Gray, A.L.; et al. Lung transplant referral for individuals with cystic fibrosis: Cystic Fibrosis Foundation consensus guidelines. J. Cyst. Fibros. 2019, 18, 321–333.
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50.
- Zhang, P.; Cheng, J.; Zou, S.; D’Souza, A.D.; Koff, J.L.; Lu, J.; Lee, P.J.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat. Commun. 2015, 6, 6221.
- Ge, Q.; Moir, L.M.; Black, J.L.; Oliver, B.G.; Burgess, J.K. TGFβ1 induces IL-6 and inhibits IL-8 release in human bronchial epithelial cells: The role of Smad2/3. J. Cell. Physiol. 2010, 225, 846–854.
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-126 Is Downregulated in Cystic Fibrosis Airway Epithelial Cells and Regulates TOM1 Expression. J. Immunol. 2010, 184, 1702–1709.
- Stolzenburg, L.R.; Wachtel, S.; Dang, H.; Harris, A. miR-1343 attenuates pathways of fibrosis by targeting the TGF-β receptors. Biochem. J. 2016, 473, 245–256.
- Zhong, T.; Perelman, J.M.; Kolosov, V.P.; Zhou, X. MiR-146a negatively regulates neutrophil elastase-induced MUC5AC secretion from 16HBE human bronchial epithelial cells. Mol. Cell. Biochem. 2011, 358, 249–255.
- Ribeiro, C.M.P.; Boucher, R.C. Role of Endoplasmic Reticulum Stress in Cystic Fibrosis–Related Airway Inflammatory Responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394.
- Oglesby, I.K.; Agrawal, R.; Mall, M.A.; McElvaney, N.G.; Greene, C.M. miRNA-221 is elevated in cystic fibrosis airway epithelial cells and regulates expression of ATF6. Mol. Cell. Pediatr. 2015, 2.
- Weldon, S.; McNally, P.; McAuley, D.F.; Oglesby, I.K.; Wohlford-Lenane, C.L.; Bartlett, J.A.; Scott, C.J.; McElvaney, N.G.; Greene, C.M.; McCray, P.B.; et al. miR-31 dysregulation in cystic fibrosis airways contributes to increased pulmonary cathepsin S production. Am. J. Respir. Crit. Care Med. 2014, 190, 165–174.
- Brown, R.; Nath, S.; Lora, A.; Samaha, G.; Elgamal, Z.; Kaiser, R.; Taggart, C.; Weldon, S.; Geraghty, P. Cathepsin S: Investigating an old player in lung disease pathogenesis, comorbidities, and potential therapeutics. Respir. Res. 2020, 21, 111.

