Cystic fibrosis (CF) is a lifelong disorder affecting 1 in 3500 live births worldwide. It is a monogenetic autosomal recessive disease caused by loss-of-function mutations in the gene encoding the chloride channel cystic fibrosis transmembrane conductance regulator (CFTR), the impairment of which leads to ionic disequilibria in exocrine organs.
- cystic fibrosis,genetic disease
- microRNA
- CFTR
1. Introduction
The dual role of time, as a fearsome enemy or important ally in the course of human diseases, has motivated researchers to advance in the frantic (and necessary) understanding of their cellular and molecular bases, hoping to identify novel approaches for patient care. The discovery of a variety of disease-modulatory molecules, including non-coding RNAs, that are present not only at the tissue level, but also in the bloodstream, has revolutionized the world of clinical molecular biology.
microRNAs (miRNAs), a class of small non-coding RNAs, are well known to regulate a diverse array of biological processes, such as proliferation, development, and metabolism [1,2]. They act as repressors of one or several genes by binding to complementary sites within the 3’ untranslated region (UTR) of target mRNAs [1,3,4].
miRNAs positively or negatively impact on the pathogenesis and/or progression of human diseases. They are (de)regulated by different mechanisms, such as epigenetic alterations, chromosomal abnormalities, as well as changes in transcriptional control [2,5]. Moreover, the altered abundance of miRNAs, along with their high stability (due to their small length of about 18–22 nucleotides) renders them highly potential biomarker candidates in non-invasive sources (i.e., blood, serum, urine, and saliva) [6,7].
The clinical potential of miRNAs as diagnostic and prognostic, as well as predictive biomarkers, has been widely described, especially in the context of human cancer [6,8,9,10]. Moreover, potential therapeutics harness the deregulation of miRNAs in disease conditions by means of miRNA mimics, so-called antimiRs [11,12].
Accumulating evidence suggests the involvement of miRNAs also in genetic diseases, including cystic fibrosis (CF) [13,14,15,16,17,18,19]. In particular, many studies showed the direct or indirect regulatory impact of deregulated miRNAs on the expression of the cystic fibrosis transmembrane conductance regulator (CFTR) mRNA [20]. Moreover, their participation in the control of the inflammation of CF airways has been demonstrated, along with their potential role as circulating biomarkers [17]. Thus, the contribution of miRNAs to disease progression and severity opens new and interesting scenarios for the clinical management of CF patients.
2. Cystic Fibrosis
2.1. Cystic Fibrosis, A Multisystemic Deadly Disease
2.2. The Mutational Landscape of the Cystic Fibrosis
Since the discovery of the first CFTR mutation in 1989, to date, more than 2000 different genetic variants have been identified (www.CFTR2.org). However, only a few of them (about 350 mutations) are considered as pathogenic (www.CFTR2.org) [43].
Severe mutations belonging to classes I, II, and III (Table 1) [27] lead to the loss of the CFTR function. In particular, class I mutations abolish the synthesis of the protein. They include nonsense mutations (i.e., premature termination codons). Class II mutations cause the retention of a misfolded protein at the endoplasmic reticulum and its subsequent degradation by the proteasome. The most common mutation in Europe, the F508del, which consists in the deletion of a phenylalanine at position 508, belongs to this class. Class III mutations affect the regulation and gating of the CFTR channel.
By contrast, class IV to VI mutations confer a milder CF phenotype (Table 1) [27]. Class IV mutations decrease the conductance of chloride and bicarbonate ions. Class V mutations lead to a reduction of the abundance (synthesis or maturation) of the normal CFTR protein. Class VI mutations destabilize the protein at the cell surface by increasing the endocytosis and lysosomal degradation of CFTR.
A new categorization of CFTR mutations into VII classes according to the therapeutic strategies has been suggested by De Boeck and Amaral [46]. The only difference compared to the traditional classification system consists in the addition of class VII mutations, which have the same functional characteristics as class I mutations but cannot be rescued by pharmacological interventions [46].
3. microRNAs as Regulators of Inflammation in Cystic Fibrosis
Inflammation plays a critical role in the progression of CF [68]. As described before, the overproduction of hyperviscous mucus obstructs airways and impairs mucociliary clearance. This creates a perfect nutrient-rich environment for bacterial colonization as well as for pro-inflammatory modulators that lead to the progressive structural damage of the lung (Figure 1).
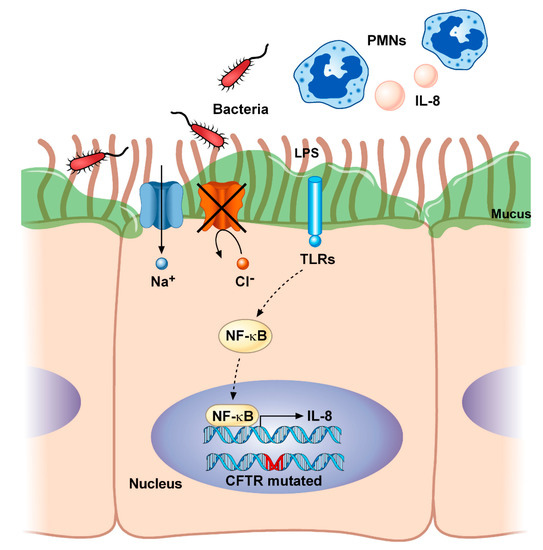
miRNAs can also control other signaling pathways to trigger inflammation in CF. High levels of miR-199a-5p were detected in human and murine CF macrophages and murine CF lungs [81]. The aberrant expression of miR-199a-5p, mediated by PI3K/AKT signaling, reduced the expression of caveolin 1 (CAV1), which is a mediator of inflammation processes, and in turn increased Toll-like receptor (TLR) 4. Moreover, the downregulation of miR-199a-5p was able to reduce the inflammation in CF macrophages via restoring the expression of CAV1. Furthermore, in vitro and in vivo studies demonstrated that the administration of the non-steroidal anti-inflammatory drug celecoxib rescued the altered pathway mediated by AKT/miR-199a-5p/CAV1 in CF macrophages and reduced lung inflammation in CFTR-deficient mice, respectively [81].
An elevated expression of miR-145, found in nasal airway cells from CF patients when compared to non-CF controls, correlated with the downregulation of SMAD3 [82]. SMAD3 is a negative modulator of the NF-kB–IL-8 pathway mediated by TGF-β1; therefore, miR-145 may contribute to CF-related inflammation [82].
Differently from the aforementioned miRNAs, miR-126 has an anti-inflammatory role in CF. It has been the first miRNA identified as deregulated in CF in 2010 [83]. In particular, low levels of miR-126 were found in CF airway epithelial cells. This downregulation correlated with the upregulation of the target of Myb protein 1 (TOM1), which is a negative regulator of TLR2, TLR4, IL-1, IL-1β, TNF-α, and NF-kB. When CF cells were stimulated with LPS and IL-1β, the knockdown of TOM1 significantly increased the NF-kB-mediated IL-8 secretion [83]. In addition, another study reported that miR-1343 attenuated the pathway of fibrosis by decreasing levels of activated TGF-β effector molecules, including phosphorylated (p) SMAD3 (pSMAD3) as well as pSMAD2 and consequently disturbing the cell migration and epithelial–mesenchymal transition [84].
Some miRNAs may also participate in chronic inflammation by affecting the remodeling of the pulmonary epithelium or by impacting the expression of genes involved in mucus hypersecretion. The role of miR-146a in CF inflammation has been linked to its negative impact on the production of the mucin 5AC (MUC5AC), which is a major component of the airway mucus [85]. Moreover, the knockdown of miR-146a in human bronchial epithelial cells resulted in the activation of the NF-kB and Jun N-terminal kinase (JNK) pathways [85].
In bronchial epithelial cells, miR-145, miR-494, and particularly miR-221 regulate the transcription factor 6 (ATF6), which is implicated in the airway inflammation through endoplasmic reticulum stress [86,87].
The deregulation of miR-31 enhanced the production of the cathepsin S (CTSS) via the direct inhibition of the interferon regulatory factor 1 (IRF-1) in CF epithelial cells [88]. CTSS is an elastinolytic and collagenolytic cysteine protease. High levels of CTSS in CF patients increased pulmonary neutrophilic infiltration of the lung, as well as the inactivation of antimicrobial proteins, such as lactoferrin and members of the β-defensin family, therefore contributing to lung inflammation and infection, and determining lung damage [89].
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics10121102