 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ana María Sánchez-Pérez | + 2226 word(s) | 2226 | 2020-11-25 09:46:49 |
Video Upload Options
A quick overview of some of the characterisitics of AD, etiology and potential treatment strategies, beyond the amyloid hypothesis.
1. Introduction
Alzheimer’s disease (AD) is the most common type of dementia, characterized by a progressive loss of memory, visuospatial and complex cognitive abilities, such as language and reasoning, which ultimately lead to a total inability to perform any type of daily activity [1][2]. Oftentimes, the patient presents comorbid psychopathologies, including depression, psychosis, anxiety, aggressive and antisocial behavior. Histologically, AD has been traditionally described by the appearance of neurofibrillary tangles (NFTs) and amyloid plaques [3]. NFTs are the intracellular aggregation of hyperphosphorylated Tau, a microtubule-associated protein that provides axonal cytoskeleton stability. Under pathological conditions (e.g., neuroinflammation and insulin resistance), Tau undergoes hyperphosphorylation, and consequently, conformational changes that reduce its affinity for microtubules [3], leading to neurodegeneration [4]. Misfolded Tau can spread via migration to neighbor healthy neurons, worsening the condition [4][5]. Senile amyloid plaques are formed by amyloid β (Aβ) peptide accumulation [6]. The old amyloid hypothesis formulated to explain AD postulates that soluble Aβ oligomers, and Aβ deposits in plaques, together with NFTs are ultimately responsible for neuronal death.
Aβ-peptide is generated from the amyloid protein precursor (APP) proteolysis. APP is a transmembrane glycoprotein expressed in a wide variety of cells and located on chromosome 21 (21q21.3, in mammals) and it undergoes proteolysis by secretases in two possible pathways (see Figure 1). The amyloidogenic pathway starts by the action of the β-site APP-cleaving enzyme 1 secretase (BACE 1), followed by the action of the γ-secretase (presenilin). This sequential cleavage releases different lengths of Aβ peptides (39–42aa), depending on γ-secretase action. Long peptides (Aβ42) are more prone to aggregation. The non-amyloidogenic pathway starts by the α-secretase action followed, as above, by γ-secretase, where no pathological peptides are generated. In healthy conditions, both pathways would compete in APP proteolysis [7]The clearance of amyloid products is carried out by microglia, the resident brain macrophages. An imbalance between the formation and clearance of Aβ peptides would result in their aggregation and accumulation in amyloid plaques [8].
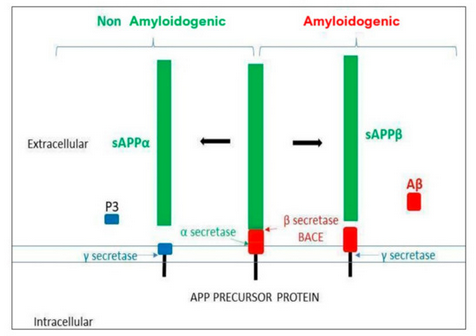
Figure 1. Amyloid precursor protein (APP) processing. The α and γ secretases are involved in the non-amyloidogenic pathway, whereas the β and the γ secretases are involved in the amyloidogenic pathway, generating the Aβ toxic oligomer.
Soluble Aβ oligomers can also be neurotoxic, since they induce intracellular oxidative stress and synaptic dysfunction [9][10] through the aberrant interaction with numerous receptors (NMDA, AMPA, acetylcholine, insulin, BDNF and receptors for advanced glycosylation end products; for a review, see [6]). Since deposition of Aβs in amyloid plaques have been observed in other dementias and in non-demented aged people, it is nowadays only considered a specific AD hallmark, unless it is observed togehter withother signs, such as NFTs, neurodegeneration, insulin resistance and neuroinflammation [11][12]. Importantly, inflammation and insulin resistance start the pathological process years before the appearance of AD’s first clinical symptoms [13][14][15].
2. Neuroinflammation in AD
Neuroinflammation is a process regulated by the brain resident macrophages, the microglia cells, which are required to recognize and eliminate any toxic component in the central nervous system (CNS) (for a review, see [16]). Microglia has a high capacity for mobility, and they can switch between two different phenotypes, M1 and M2, that are characterized by a different morphology and cytokine expression profile. The M2 phenotype is the “resting” type that actively monitors the brain in healthy conditions [17]. The switch to M1 begins with the recognition of the pathogen-associated molecular patterns (PAMPs) or the damage-associated molecular patterns (DAMPS) by the pattern recognition receptors (PRRs). These include the ‘toll-like receptors’ (TLRs) in microglia membrane (both plasma and endosomal membrane), the cytoplasmic NOD-like receptors (NLR), the intracellular retinoic acid-inducible gene-I-like receptors and the transmembrane C-type lectin receptors (for a review, see [18]). PAMPS and DAMPS range from bacterial wall components, lectins, lipopolysaccharides (LPS) and virus capsid proteins, to debris released by dying cells and Aβ oligomers [19]. The activation of this system, the so-called inflammasome, initiates the inflammatory cascade, that will induce the secretion of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and interleukins 1β, 6 and 18 (IL-1β, IL-6 and IL-18, respectively). Pro-inflammatory cytokines purpose is to orchestrate the neutralization and elimination of toxic molecules and cellular debris. In normal conditions, once the toxic stimuli have been cleared, microglia swifts to the anti-inflammatory (M1) phenotype and secretes anti-inflammatory cytokines such as interleukins 4, 10 and 18 (IL-4, IL-10 and IL-18, respectively), brain-derived neurotrophic factor (BDNF) or nerve growth factor (NGF), whose role is to terminate the innate immune response and contribute to restore the synaptic function. However, under pathological conditions, microglia cells do not go back to their resting state, thus causing a chronic inflammation process, with the overproduction of pro-inflammatory cytokines and reduction of neuroprotective factors that in sustained situations become highly toxic, leading to neurodegeneration [20].
Therefore, the chronic neuroimmune system activation underlies the initiation and progression in many dementias, and it is involved in the late onset of AD [21][22][23]. Not only Aβ activates the microglia [24], but also misfolded Tau interaction with microglia triggers inflammation [25]. The elimination of the microglial receptor, NLR family pyrin domain containing 3 (NLRP3) has shown to reduce brain Aβ levels in rodent models of AD [26][27]; since then, NLRP3 inflammasome has been deeply studied and characterized in AD [28][29]. In addition to the neurological symptoms, neuroinflammation also underlies the psychiatric signs associated with AD, and for that reason, targeting neuroinflammation has also been proposed to treat those comorbid disturbances [30].
According to the neuroinflammation hypothesis underlying AD, there is a lower incidence of AD among users of chronic non-steroidal anti-inflammatory molecules (NSAIDs) [31][32]. NSAIDs inhibit mostly the cyclooxygenase (COX) activity, which synthesizes prostaglandin (PG) from arachidonic acid. At least two isoforms have been described, COX-1 and 2. COX-1 is expressed constitutively; in contrast, COX-2 is induced by inflammation and cellular stress, increasing PG production [33]. Anti-inflammatory compounds, inhibiting COX activity, Naproxen and Celecoxib have been tested in clinical trials against AD. Naproxen, a non-selective COX inhibitor was administered (220 mg/twice day for two years) to 195 pre-symptomatic AD subjects (aged 55+) with a familial history of AD. The progression of the disease was evaluated with the Alzheimer’s Progression Score (APS). Although Naproxen reduced the rate of the APS, the data was not significant [34]. Celecoxib, a selective COX-2 inhibitor, was administrated (200 mg/twice day for 2 years) in 677 pre-symptomatic subjects (70+) with at least one first-degree relative with AD. No improvement in the cognitive symptoms in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT) in the AD patients compared to the placebo group was found [35]. None of these clinical trials analyzed inflammation biomarkers; therefore, these studies cannot test the neuro-inflammation hypothesis underlying AD progression. In addition, another conclusion from these clinical data is to shift the focus to different inflammation pathways, other than the COX-PG pathway.
For example, the specific TNF-α inhibitor, Etanercept, was evaluated in a small group of 41 AD patients (55+) with mild to severe AD (SMMSE score between 10 and 27), to test its anti-inflammatory effect and subsequent improvement of cognitive function. The weekly 50 mg subcutaneous administration was well tolerated; however, after 24 weeks of treatment, Etanercept did not show significant beneficial effects in cognition, behavior, systemic cytokine levels or global function compared to the placebo-treated group [36]. The failure of this clinical trial, maybebe due to i) Also, the period of time of assays is too short; and/or ii) there are other factors that may be involved for instance insulin resistance [37]; thus, inhibiting specifically the TNF-α action may not be sufficient to counteract the inflammasome activity.
3. Targeting Insulin Resistance to Treat AD
The late onset of AD is strongly associated with insulin resistance; in fact, AD has been often recognized as Type 3 diabetes [38]. Several situations can bring about insulin resistance: metabolic syndrome caused by high fat diet, sedentarism, obesity, genetic predisposition and neuroinflammation [39]. Insulin resistance increases Tau aberrant phosphorylation, the expression of APP and the formation of Aβ oligomers and its deposition. In addition, insulin resistance augments oxidative and endoplasmic reticulum stress, mitochondrial dysfunction and pro-inflammatory cascades [40]. Not surprisingly, Type 2 Diabetes mellitus (T2DM) has been associated with cognitive impairment [41].
For this reason, administration of intranasal (IN) insulin has been considered as a potential therapeutic strategy against AD. A systematic review on this strategy concluded that whereas IN insulin administration showed improvement in verbal memory and story recall, it was not effective on other aspects of cognition. Interestingly, the authors conclude that the treatment is affected by the presence of Apoe4 isoform, where Apoe4 (–) patients displayed more benefits compared to Apoe4 (+) patients. This systematic review concluded that IN insulin (although very safe, not interfering with systemic glucose levels) does not seem an effective treatment for dementia associated to AD or mild cognitive impairment (MCI). Most importantly, these data support that proper stratification by disease stage, Apoe4 carrier status and different types of insulin must be considered for a better therapeutic effect [42].
In Insulin resistance situations, the administration of insulin will not effective in the long term; thus, other treatments have been developed to enhance the insulin sensitivity instead, These strategies include the activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK). AMPK activation inhibits the mammalian target of rapamycin (mTOR)/p70 ribosomal S6 kinase (p70S6K) activity [43]. The mTOR/p70S6K pathway is activated by insulin and phosphorylates the insulin receptor substate 1 (IRS1) on serine residues as a negative feedback loop to reduce insulin signaling [44][45] (see Figure 2). Interestingly, AMPK activity displays an anti-inflammatory effect, decreasing inflammatory cells proliferation and their adhesion to the blood vessel endothelium. AMPK activity also reduces amyloidogenesis, Tau hyperphosphorylation and the activation of autophagic degradation[43]. In agreement with this, a pilot study in non-diabetic subjects (aged 55–80 years) diagnosed with MCI, Metformin (an AMPK activator) administration, ameliorated learning, memory and attentional abilities, evaluated by the Paired Associates Learning (PAL) scale and DMS Percent Correct Simultaneous. Interestingly, there was the improvement in behavior, but no changes in the cerebrospinal fluid (CSF) of Aβ42, and total or phosphorylated Tau levels were found [46], further suggesting the idea that the amyloid hypothesis does not accurately explain AD.
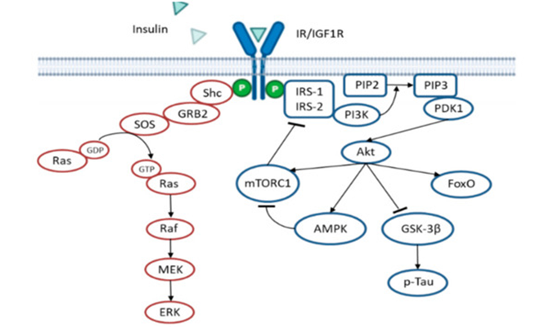
Figure 2. Insulin signaling cascade. The scheme shows the negative feedback mechanism that mTORC1 exerts over IRS1/2. Activation of AMPK inhibits mTORC1, thus improving insulin signaling. In pathological situations, insulin resistance reduces Akt activity, leading to higher GSK-3β activity and subsequent Tau hyperphosphorylation, an important hallmark of AD.
Milk-derived proteins have also been proposed as possible antioxidant and anti-inflammatory compounds, given their capability to reduce insulin resistance. For example, lactoferrin (a multifunctional iron-binding glycoprotein) administration increases insulin sensitivity in adipose tissue explants from obese subjects [47][48]. Indeed, Lactoferrin antioxidant function is highly dependent on its iron binding capacity [49]. In metabolic syndrome, iron accumulation is considered an important factor underlying insulin resistance and oxidative stress; accordingly, iron-chelators have a positive effect ameliorating the physiopathology of obesity. Lactoferrin therapeutic potential against AD was tested in a pilot study with AD patients. Short-term administration of lactoferrin (250 mg/day for three months) reduced serum oxidative levels and neuroinflammatory markers, and regulated neurotransmitters serum levels concomitant with improved cognitive performance, compared to control [50].
Moreover, deficiency in micronutrients such as vitamin B12 (critical for mental health [51]) has been associated with insulin resistance [51][52][53]. Interestingly, combined treatment of folic acid and vitamin B12 has been shown to improve AD cognitive performance in a randomized trial of 240 patients diagnosed with MCI for 6 months, concomitant with a reduction in serum inflammatory markers [54]. Additionally, Vitamin B12 in combination with anti-psychotic drugs (Risperidone and Quetiapine) reduced blood levels of the pro-inflammatory cytokines IL-8 and TNF-α and augmented the expression of the anti-inflammatory cytokine TGF-β, compared to non-treated AD patients [55]. The same medication formulation was tested in psychotic patients for the expression of the Cluster of Differentiation 68 (CD68), a protein expressed by monocytes and macrophages that has been shown to correlate positively with psychotic symptoms in AD patients. This treatment reduced CD68 expression [56], and therefore, has been proposed as a good strategy against AD. In addition, CD68 has been shown to bind and internalize oxidized Low-Density Lipoprotein (oxLDL), a cholesterol carrier [57], suggesting a relationship of CS68 with intracellular lipid accumulation and atherogenesis.
In this line of research, pharmacological treatments used to treat other diseases, such as hypertension (i.e., calcium channel blockers) [58][59] or hypercholesterolemia (i.e., statins) [60][61], were postulated as therapeutic agents against AD, given their alleged anti-inflammatory and insulin sensitizing properties. The results from a randomized clinical trial demonstrated that, for instance, the calcium channel blocker nilvadipine has no beneficial effects in a clinical trial against treating AD [62]. On the other hand, statins’ potential therapeutic effect against Alzheimer seems controversial. Simvastatin has been shown to improve memory deficits only at higher doses (80 mg/daily for 18 months) in small groups of patients (50+) [63]; lower doses in larger groups, even though they were efficient in lowering lipid levels, did not ameliorate memory performance [64]. Another statin has been evaluated, artovastatin. In an 18 months clinical trial in dyslipidemic patients, although artovastatin effectively corrected dyslipidemia and inflammatory markers, cognitive function was not evaluated in the study [65]; furthermore, a randomized clinical trial demonstrated no beneficial effects of artovastatin treatment on AD patients’ symptoms [66]. These data conclude that although there is a promising therapeutic evidence in correcting dyslipidemia in AD, more studies are needed to establish statins therapeutic applications in AD patients, since reasonable concerns arises with lowering of the cholesterol molecule itself
In conclusion, the clinical data reviewed suggest a promising potential to treat neuroinflammation in AD to ameliorate the symptoms. Further studies are required to establish an effective treatment protocol to address this complex disease.
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429.
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A Review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 1–15.
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732.
- Szabo, L.; Eckert, A.; Grimm, A. Insights into disease-associated tau impact on mitochondria. Int. J. Mol. Sci. 2020, 21, 6344.
- Vogel, J.W.; Initiative, A.D.N.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; The Swedish BioFINDER Study. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 1–15.
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 2012, 33, S67–S78.
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S.S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270.
- Lee, C.Y.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960.
- Birla, H.; Minocha, T.; Kumar, G.; Misra, A.; Singh, S.K. Role of oxidative stress and metal toxicity in the progression of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 552–562.
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. PubMed. Oxid. Med. Cell. Longev. 2013, 2013, 1–10.
- Skaper, S.D. Alzheimer’s disease and amyloid: Culprit or coincidence? Int. Rev. Neurobiol. 2012, 102, 277–316.
- Lee, K.S.; Chung, J.H.; Choi, T.K.; Suh, S.Y.; Oh, B.H.; Hong, C.H. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 281–287.
- DaRocha-Souto, B.; Scotton, T.C.; Coma, M.; Serrano-Pozo, A.; Hashimoto, T.; Serenó, L.; Rodríguez, M.; Sánchez, B.; Hyman, B.T.; Gómez-Isla, T. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 2011, 70, 360–376.
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966.
- Tarkowski, E.; Andreasen, N.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–120.
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629.
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090.
- Albornoz, E.A.; Woodruff, T.M.; Gordon, R. Inflammasomes in CNS Diseases. Experientia Supplementum 2018, 108, 41–60.
- Bagyinszky, E.; Van Giau, V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254.
- Obulesu, M.; Lakshmi, M.J. Neuroinflammation in Alzheimer’s disease: An understanding of physiology and pathology. Int. J. Neurosci. 2013, 124, 227–235.
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Valentina, G.; Millet, P. In vivo TSPO signal and neuroinflammation in Alzheimer’s disease. Cells 2020, 9, 1941.
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nat. Cell Biol. 1995, 374, 647–650.
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s disease in the context of Tau pathology. Biomolecules 2020, 10, 1439.
- François, A.; Bilan, A.R.; Quellard, N.; Fernandez, B.; Janet, T.; Chassaing, D.; Paccalin, M.; Terro, F.; Page, G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflamm. 2014, 11, 1–14.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678.
- Hanslik, K.L.; Ulland, T.K. The Role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front. Neurol. 2020, 11, 11.
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 101192.
- Clement, A.; Wiborg, O.; Asuni, A.A. Steps towards developing effective treatments for neuropsychiatric disturbances in Alzheimer’s disease: Insights from preclinical models, clinical data, and future directions. Front. Aging Neurosci. 2020, 12, 56.
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J. Alzheimer’s Dis. 2016, 54, 853–857.
- Pasinetti, G.M. From epidemiology to therapeutic trials with anti-inflammatory drugs in Alzheimer’s disease: The role of NSAIDs and cyclooxygenase in β-amyloidosis and clinical dementia1. J. Alzheimer’s Dis. 2002, 4, 435–445.
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000.
- Breitner, J.; Meyer, P.-F. Author response: INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2020, 94, 594.
- Breitner, J.; Baker, L.; Drye, L.; Evans, D.; Lyketsos, C.G.; Ryan, L.; Zandi, P.; Saucedo, H.H.; Anau, J.; Cholerton, B. Follow-up evaluation of cognitive function in the randomized Alzheimer’s disease anti-inflammatory prevention trial and its follow-up study. Alzheimer’s Dement. 2015, 11, 216–225.
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.L.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168.
- Najem, D.; Bamji-Mirza, M.; Chang, N.; Liu, Q.Y.; Zhang, W. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev. Neurosci. 2014, 25, 509–525.
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1078–1089.
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 1–9.
- De La Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimers disease. Curr. Alzheimer Res. 2012, 9, 35–66.
- Cukierman, T.; Gerstein, H.C.; Williamson, J.D. Cognitive decline and dementia in diabetes-systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469.
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.-A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510.
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474.
- Hartley, D.; Cooper, G.M. Role of mTOR in the degradation of IRS-1: Regulation of PP2A activity. J. Cell. Biochem. 2002, 85, 304–314.
- Yoneyama, Y.; Inamitsu, T.; Chida, K.; Iemura, S.-I.; Natsume, T.; Maeda, T.; Hakuno, F.; Takahashi, S.-I. Serine phosphorylation by mTORC1 promotes IRS-1 degradation through SCFβ-TRCP E3 ubiquitin ligase. iScience 2018, 5, 1–18.
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113.
- Fernández-Real, J.M.; García-Fuentes, E.; Moreno-Navarrete, J.M.; Murri-Pierri, M.; Garrido-Sánchez, L.; Ricart, W.; Tinahones, F. Fat overload induces changes in circulating lactoferrin that are associated with postprandial lipemia and oxidative stress in severely obese subjects. Obesity 2010, 18, 482–488.
- Artym, J. A remedy against obesity? The role of lactoferrin in the metabolism of glucose and lipids. Postęp. Higien. Med. Dośw. 2012, 66, 937–953.
- Brizzio, E.; Castro, M.; Narbaitz, M.; Borda, N.; Carbia, C.D.; Correa, L.; Mengarelli, R.; Merelli, A.; Brizzio, V.; Sosa, M.; et al. Ulcerated hemosiderinic dyschromia and iron deposits within lower limbs treated with a topical application of biological chelator. Veins Lymphat. 2012, 1, e6.
- Mohamed, W.A.; Salama, R.M.; Schaalan, M.F. A pilot study on the effect of lactoferrin on Alzheimer’s disease pathological sequelae: Impact of the p-Akt/PTEN pathway. Biomed. Pharmacother. 2019, 111, 714–723.
- Manzanares, W.; Hardy, G. Vitamin B12: The forgotten micronutrient for critical care. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 662–668.
- Kouroglou, E.; Anagnostis, P.; Daponte, A.; Bargiota, A. Vitamin B12 insufficiency is associated with increased risk of gestational diabetes mellitus: A systematic review and meta-analysis. Endocrine 2019, 66, 149–156.
- Li, Z.; Gueant-Rodriguez, R.-M.; Quilliot, D.; Sirveaux, M.-A.; Meyre, D.; Gueant, J.-L.; Brunaud, L. Folate and vitamin B12 status is associated with insulin resistance and metabolic syndrome in morbid obesity. Clin. Nutr. 2018, 37, 1700–1706.
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of folic acid and vitamin B12, alone and in combination on cognitive function and inflammatory factors in the elderly with mild cognitive impairment: A single-blind experimental design. Curr. Alzheimer Res. 2019, 16, 622–632.
- Vakilian, A.; Razavi-Nasab, S.M.; Ravari, A.; Mirzaei, T.; Moghadam-Ahmadi, A.; Jalali, N.; Bahramabadi, R.; Rezayati, M.; Yazdanpanah-Ravari, A.; Bahmaniar, F.; et al. Vitamin B12 in association with antipsychotic drugs can modulate the expression of pro-/anti-inflammatory cytokines in Alzheimer disease patients. Neuroimmunomodulation 2018, 24, 310–319.
- Bahramabadi, R.; Samadi, M.; Vakilian, A.; Jafari, E.; Fathollahi, M.S.; Arababadi, M.K. Evaluation of the effects of anti-psychotic drugs on the expression of CD68 on the peripheral blood monocytes of Alzheimer patients with psychotic symptoms. Life Sci. 2017, 179, 73–79.
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838.
- Farah, R.R.; Khamisy-Farah, R.S.-S. Calcium channel blocker effect on insulin resistance and inflammatory markers in essential hypertension patients. PubMed. Int. Angiol. 2013, 32, 85–93.
- Cabezas-Cerrato, J.; García-Estévez, D.A.; Araújo, D.; Iglesias, M. Insulin sensitivity, glucose effectiveness, and β-cell function in obese males with essential hypertension: Investigation of the effects of treatment with a calcium channel blocker (diltiazem) or an angiotensin-converting enzyme inhibitor (quinapril). Metabolism 1997, 46, 173–178.
- Weitz-Schmidt, G. Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 2002, 23, 482–487.
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34.
- Lawlor, B.A.; Segurado, R.; Kennelly, S.; Rikkert, M.G.M.O.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018, 15, e1002660.
- Huang, W.; Li, Z.; Zhao, L.; Zhao, W. Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of Alzheimer’s disease via modulating the expression of miR-106b. Biomed. Pharmacother. 2017, 92, 46–57.
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; Van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011, 77, 556–563.
- Zhao, L.; Zhao, Q.; Zhou, Y.; Zhao, Y.; Wan, Q. Atorvastatin may correct dyslipidemia in adult patients at risk for Alzheimer’s disease through an anti-inflammatory pathway. CNS Neurol. Disord. Drug Targets 2016, 15, 80–85.
- Feldman, H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; Demicco, D.A.; et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 2010, 74, 956–964.

