 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Kalamvoki | + 8574 word(s) | 8574 | 2020-12-30 05:27:51 | | | |
| 2 | Rita Xu | -4976 word(s) | 3598 | 2021-01-11 06:50:43 | | |
Video Upload Options
Approximately half of the proteins encoded by the herpes simplex virus 1 (HSV-1) genome have been classified as non-essential. These proteins have essential roles in vivo in counteracting antiviral responses, facilitating the spread of the virus from the sites of initial infection to the peripheral nervous system, where it establishes lifelong reservoirs, virus pathogenesis, and other regulatory roles during infection.
1. Introduction
The large family of DNA viruses, Herpesviridae, have co-evolved with mammals for millions of years [1][2]. The family Herpesviridae is further divided into three subfamilies, Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae. Herpes simplex virus type 1 (HSV-1), a member of Alphaherpesvirinae, is one of the most well-studied representatives of this family of viruses and will be the focus of this review.
HSV-1 is an enveloped dsDNA virus, which has a genome size of about 152 kb and virion size of about 150–300 nm in diameter [3]. The virion contains the envelope decorated with viral glycoproteins and a proteinaceous layer known as the tegument, which surrounds the capsid of the virus containing the genome. HSV-1 is an important human pathogen, with approximately 80% of the human population infected [3]. Symptoms of HSV-1 infection vary, from lesions in the oral-facial region (“cold sores”), to herpes keratitis, the leading cause of infectious blindness, to herpes encephalitis, which can be fatal. When HSV-1 encounters a host, it will first infect the mucosal epithelial cells in the oral-facial region (although HSV-1 also causes genital infections). It is in these cells that the virus undergoes lytic replication. The virus will then infect innervating sensory neurons, travel anterograde to the trigeminal ganglia (TG), and establish latency, where it will remain for the life of the infected individual. When HSV-1 undergoes latency there are very few genes expressed, including an 8.3-kb region known as the latency-associated transcript (LAT), which is a long non-coding regulatory RNA spliced to about 1.5- and 2-kb introns that have regulatory roles on viral genes expression, and a 6.3-kb exon encoding multiple microRNAs, which target many of the IE genes and other lytic genes, thus suppressing viral replication [4][5][6][7][8][9][10]. It seems that LAT may be important for reactivation of HSV-1 from latency and for blocking apoptosis [11][12][13][14][15][16][17][18]. Periodically, HSV-1 will reactivate from latency due to stress, immunosuppression, or other stimuli, and newly produced virions will travel retrograde to the initial site of infection. There is currently no cure for HSV-1 and no vaccine.
There are three classes of viral genes for HSV-1 and they are expressed in a cascade fashion [19][20]. The virus first encodes the immediate-early (IE) or alpha (α) genes (ICP0, ICP4, ICP27, ICP22, or ICP47) whose products are important for expression of the next class of viral genes, the early (E) or beta (β) genes. The early genes encode proteins largely involved in viral DNA replication and, along with the immediate-early genes, facilitate the expression of the late class of viral genes. The late (L) or gamma (γ) class of viral genes express proteins involved in virion assembly and egress. HSV-1 genes are also divided based on stretches of unique sequences in the genome. Therefore, there are a class of unique long (UL) and unique short (US) genes depending on which region of the genome the gene is expressed from. These unique regions are flanked by inverted repeats. Thus, the HSV-1 genome is structured as follows: TRL-UL -IRL-IRS-US-TRS. There are about 58 UL genes and about 13 US genes that have been characterized for functionality, though there are more viral genes that have not been well characterized or described (Dolan 1998).
Interestingly, although HSV-1 is known to encode 80 genes, it has also been found that about half of these genes are non-essential for viral replication in cell culture [21][22]. Essential genes of HSV-1 are involved in viral DNA replication, the transcription of certain viral genes, genes encoding capsid proteins, genes encoding viral DNA packaging proteins, and some envelope glycoproteins. HSV-1 genes determined to be non-essential are involved in nucleic acid metabolism, combating various host responses to infection, facilitating optimal viral replication, facilitating primary envelopment, virus pathogenesis, or other functions that are not yet characterized (Table 1). While deletion of the non-essential genes in cell culture does not inhibit viral replication, these genes are generally essential for replication in the natural human host as mutant viruses deleted of non-essential genes have rarely been isolated from a patient. One example are mutants in the viral glycoprotein gC that have been recovered from patients with recurrent herpes keratitis [23][24].
Table 1. Non-essential genes of HSV-1, corresponding proteins, their location on the HSV-1 virion, and their function. Pink: tegument proteins, blue: accessory proteins, yellow: envelope proteins, green: capsid proteins.
|
Gene |
Protein |
Location on Virion |
Function |
|
RL1 or γ134.5 |
ICP34.5 |
tegument |
Prevents host translational shutoff and autophagy |
|
RL2 or α0 |
ICP0 |
tegument |
Promiscuous transactivator of genes, disrupts repressor complexes, E3 ubiquitin ligase, inhibits innate immunity, modulates endocytosis, etc. |
|
UL2 |
uracil-DNA glycosylase |
accessory |
nucleic acid metabolism |
|
UL3 |
|
accessory |
|
|
UL4 |
|
accessory |
|
|
UL7 |
|
tegument |
Virion assembly and egress |
|
UL10 |
gM |
envelope |
Host and viral protein trafficking |
|
UL11 |
|
tegument |
Cytoplasmic envelopment |
|
UL12 |
|
accessory |
Nucleic acid metabolism |
|
UL12.5 |
|
accessory |
Involved in depleting mtDNA |
|
UL13 |
Ser/thr protein kinase |
tegument |
Blocking innate immune responses, supporting viral protein synthesis |
|
UL16 |
|
tegument |
Cytoplasmic envelopment |
|
UL20 |
|
envelope |
Glycoprotein trafficking |
|
UL21 |
|
tegument |
Promotes capsid egress to the cytoplasm |
|
UL23 |
thymidine kinase (TK) |
tegument |
Broad spectrum nucleoside kinase |
|
UL24 |
|
accessory |
Glycoprotein trafficking, nucleolus dispersal |
|
UL31 |
|
accessory |
Component of the nuclear egress complex (NEC), promotes primary nuclear envelopment |
|
UL34 |
|
accessory |
Component of the nuclear egress complex (NEC), promotes primary nuclear envelopment |
|
UL35 |
VP26 |
capsid |
Affects DNA packaging, mediates capsid assembly, trafficking post viral entry |
|
UL39 |
RR1 (ribonucleotide reductase) |
accessory |
Part of the ribonucleotide reductase (RR) complex, converts ribonucleotide diphosphates to corresponding deoxyribonucleotides, allowing for virus replication particularly in non-dividing cells |
|
UL40 |
RR2 (ribonucleotide reductase) |
accessory |
Part of the ribonucleotide reductase (RR) complex, converts ribonucleotide diphosphates to corresponding deoxyribonucleotides, allowing for virus replication particularly in non-dividing cells |
|
UL41 |
VHS |
tegument |
Viral RNase, degrades host transcripts and blocks antiviral responses |
|
UL43 |
|
tegument |
|
|
UL44 |
gC |
envelope |
Mediates viral binding to heparan sulfate, regulates entry by a low-pH pathway |
|
UL45 |
|
envelope |
Required for syncytia formation during HSV-1 gB syn infection |
|
UL46 |
VP11/12 |
tegument |
Regulation of transcription, activates pathways for cell survival, blocks pathways for innate immunity activation |
|
UL47 |
VP13/14 |
tegument |
Regulation of transcription, modulating post-transcriptional processing of mRNAs |
|
UL49 |
VP22 |
tegument |
Facilitates viral gene expression, protein expression, and DNA replication; inhibits inflammasome |
|
UL49.5 |
gN |
envelope |
Binding partner of gM |
|
UL50 |
|
tegument |
Nucleic acid metabolism |
|
UL51 |
|
tegument |
Participates in cytoplasmic envelopment; facilitates virus spread from cell-to-cell; recruits UL7 to tegument |
|
UL53 |
gK |
envelope |
Participates in virion egress from host cell; regulates virus entry and fusogenic activity of the virion; complexes with UL20 |
|
UL55 |
|
tegument |
Participates in cytoplasmic envelopment |
|
UL56 |
|
tegument |
Participates in cytoplasmic envelopment |
|
US1 |
ICP22 |
accessory |
Regulates viral late gene expression; facilitates formation of complexes important for protein folding; participates in primary envelopment; blocks immune responses |
|
US1.5 |
|
accessory |
Participates in viral gene transcription |
|
US2 |
|
tegument |
Protein trafficking |
|
US3 |
Ser/thr protein kinase |
tegument |
Blocks apoptosis, enhances viral gene expression, facilitates capsid nuclear egress, phosphorylates numerous substrates |
|
US3.5 |
Ser/thr protein kinase |
tegument |
Phosphorylates substrates but cannot block apoptosis and does not facilitate nuclear egress |
|
US4 |
gG |
envelope |
Regulation of chemokines |
|
US5 |
gJ |
envelope |
Inhibits apoptosis and cell stress pathways |
|
US7 |
gI |
envelope |
Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence |
|
US8 |
gE |
envelope |
Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence |
|
US8.5 |
|
accessory |
Localizes in the nucleoli |
|
US9 |
|
tegument |
Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence |
|
US10 |
|
tegument |
Important for neurovirulence |
|
US11 |
|
tegument |
Block PKR activation and shutoff of host translation; block IFN induction; regulation of virus genes expression; trafficking of unenveloped capsids |
|
US12 |
ICP47 |
accessory |
Prevents MHC I antigen presentation, supports neurovirulence |
It is of great interest to understand the roles of non-essential genes to better understand virus–host interactions. Moreover, the non-essential genes have properties that make them attractive for the development of therapeutics. There are varying degrees of deficiency of viruses mutated for non-essential genes when grown in cell culture, and for some of these genes, the defect is cell type specific [25][26]. There is still much to learn about the non-essential genes of HSV-1. Here, we present a comprehensive analysis of the current understanding of the roles of non-essential genes of HSV-1. We explore the functions ascribed to these genes and their corresponding proteins, the potential treatment and therapeutic avenues that can be explored based on the functions and characterization of select HSV-1 non-essential genes, and the complex and intricate roles of non-essential genes in HSV-1 infection.
2. Repressors of Gene Silencing, Viral Transactivators, and Host Evasion Factors
2.1. RL2 or α0 (ICP0)
The infected cell protein 0 (ICP0) of HSV-1 was first reported as a nuclear phosphoprotein with an essential role in cell cultures only at low multiplicity of infection (MOI). ICP0 was deemed to be non-essential at high multiplicities of infection in cell cultures, but viral gene expression was reduced [19][27][28][29][30][31][32][33]. In certain cell lines, particularly cancer cell lines, such as the human osteosarcoma (U2OS), an ICP0-null virus replicates as efficiently as wild-type virus, which may be due to impaired recruitment of antiviral factors to the sites of viral gene transcription and DNA replication and/or due to lack of certain restriction factors [26][28][30][32][34][35]. Genes coding for ICP0 are present in the genomes of simplex and varicelloviruses, but they are absent from the mardivirus genus. These proteins show strong sequence homology to ICP0 within the RING (Really Interesting New Gene) finger domain. Orthologs of ICP0 are also present in lymphocryptoviruses (e.g., EBV) and the cytomegalovirus (CMV) [36][37][38]. The functions of ICP0 are broad, from activation of transcription and chromatin remodeling, to evasion of antiviral responses, cell cycle effects, interfering with DNA damage responses, and endocytosis.
In early studies, ICP0 was found with ICP4 to stimulate ICP8 expression in transfection assays [39][40]. Furthermore, it was shown to function as a potent transactivator of different genes introduced into cells by transfection or infection, including the viral thymidine kinase (TK) gene and ICP6 gene, the human immunodeficiency virus (HIV) LTR, and several human papillomavirus (HPV) genes [41][42][43][44][45][46][47][48][49][50]. In fact, ICP0 was found to stimulate the expression of all three classes of HSV genes [31][51]. Therefore, ICP0 was proposed to be a promiscuous transactivator of gene expression.
ICP0 also functions as an E3 ubiquitin ligase and most substrates ubiquitinated by ICP0 appear to be targeted for degradation (Figure 1A). This activity of ICP0 was mapped to residues 116-156, where there is a Zn2+-binding RING finger domain [52][53][54][55]. To exert its E3 ubiquitin ligase function, ICP0 forms a complex with different ubiquitin conjugation enzymes, including UbcH5a and UbcH6 [52][56][57][58][59][60]. Major targets of ICP0 are components of the nuclear domain 10 (ND10) bodies. As a DNA virus, the genome of HSV-1 transcribes and replicates in the nucleus. The host attempts to block viral gene expression and replication by entrapping the viral DNA in promyelocytic leukemia (PML)-nuclear bodies (NBs) and depositing histones and other repressor complexes on it. The main protein that orchestrates the formation of ND10 bodies is the PML. Other components of ND10s include the Sp100, Daxx, Mre 11, ATM, ATRX, p53, and others. ICP0 disrupts the ND10s by causing degradation of the different isoforms of PML, Sp100, and potentially of other proteins (Figure 1A) [34][59][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77] Notably, several components of the ND10 bodies are interferon inducible genes, which underscores the synergy between gene silencing mechanisms and innate immunity in suppressing HSV-1 gene expression. ICP0-null viruses or E3 ubiquitin ligase mutants have viral DNA entrapped in PML-NBs at low MOI and display reduced transactivation activity and ability to block antiviral responses [34][64][78][79][80][81][82][83]. ICP0 E3 ligase-deficient viruses are hypersensitive to interferon, replicate poorly, and fail to reactivate efficiently from neuronal latency [25][55][67][68][69][70][71]. Based, on these observations, Dr. Kalamvoki’s group recently developed a high-throughput assay to screen for ICP0-E3 ubiquitin ligases inhibitors [72]. This assay is proximity based and takes advantage of the fact that ICP0 is autoubiquitinated and degraded during infection and that this ICP0 autoubiquitination can occur in vitro using the purified protein encoded by the exon II of ICP0 (contains the RF domain), UbcH5a, and Ub [60][73][74]. Screening a small compound library, Dr. Kalamvoki’s group identified potential scaffolds that can interfere with the ICP0 E3 ubiquitin ligase activity [72].
ICP0 has seven SUMO-interacting motif (SIM)-like sequences (SLSs), and multiple ND10 components, including PML and SP100, are SUMOylated; therefore, ICP0 could bind to them (Figure 1A) [34][56][75][76][77][78][79][80][81]. It has been found that inhibition of cellular ubiquitination led to an increase of SUMOylated proteins that ended up accumulating at PML-NBs [82]. ICP0 utilizes both SUMO-dependent and SUMO-independent mechanisms to degrade Sp100 and multiple PML isoforms in an effort to prevent restriction of the virus by the host [34][56][75][76][78][79][83][84]. Other proteins could also be the target of SUMO-dependent degradation by ICP0 [75][77][81]. Specifically, SUMO-dependent degradation of MORC3 by ICP0, which associates with Sp100, has been observed and this occurs in a RING-finger-dependent manner and appears to diminish the association of PML-NBs with viral DNA [85]. Additionally, there has been a function ascribed to ICP0 SUMO–SIM interactions at the ND10s to modulate the DNA damage response (DDR) during infection [86][87]. For example, the DNA repair function of the DNA-dependent protein kinase (DNA-PK) is inhibited by ICP0 through degradation of its catalytic subunit and this facilitates virus replication [88][89][90]. Additionally, ICP0 mediates the degradation of two E3 ubiquitin ligases RFN8 and RFN168 that act as mediators of the ATM pathway and trigger recruitment of downstream effectors to sites of double-strand DNA breaks [91][92][93][94][95]. More work will need to be done to characterize the degradation of SUMOylated proteins by ICP0 that are not related to the ND10s. The ability for ICP0 to interrupt SUMO interactions and to degrade SUMOylated proteins during infection is likely a strategy to modify the cellular proteome to both prevent antiviral responses and promote the infection [34][76][83].
In tandem with the dispersion of ND10 bodies, ICP0 activates the viral chromatin (Figure 1B). Immediately after its release in the nucleus, HSV-1 DNA associates with repressive histones and other repressor complexes [96][97][98]. However, markers of active gene expression label the viral chromatin during lytic infection, such as tri-methylation of histone H3 at lysine 4 (H3K4) and acetylation of H3 at lysine 9 and lysine 14 [97][98][99], while suppressive epigenetic modifications of histone H3 (H3K9me3 and H3K27me3) are removed in an ICP0-dependent manner (Figure 1B) [65][100][101]. ICP0 was also found to associate with class II HDACs in vitro and control their repressor activity [102][103]. In addition, ICP0 seems to promote histone acetylation, as demonstrated using inhibitors of histone deacetylases [103][104][105]. This is also supported by the fact that ICP0 recruits to the viral genome the histone acetyltransferase CLOCK through interaction with the circadian regulator protein BMAL1. This leads to recruitment of additional viral transactivators ICP4, ICP22, ICP27, and part of the host transcription complex TFIID [106][107][108]. Tandemly, ICP0 disrupts repressor complexes, such as the REST/CoREST/HDAC complex and LSD1 [109][110][111][112]. ICP0 disperses the REST/CoREST/HDAC1/2/LSD1 through interaction with CoREST in an effort to promote HSV-1 gene expression and DNA replication (Figure 1B) [78][112][113][114][115]. It was also found that the interferon-inducible gene 16 (IFI16), Daxx, and ATRX proteins serve to restrict the virus, likely through sensing of viral DNA and obstructing replication and causing deposition of silencing histone H3 [34][65][100][116][117][118][119][120]. ICP0 induces the degradation of ATRX and IFI16 [121][122][123][124][125]. Degradation of ATRX seems to be secondary to PML degradation, while depletion of ICP0 appears to be both ICP0 dependent and independent.
ICP0 has also been shown to harness cell cycle components to support the infection. Thus, ICP0 was found to recruit cyclin D3 and the kinase cdk4 to ND10s to enable viral gene transcription and DNA replication, which was also supported by the fact that ICP0 nuclear-to-cytoplasmic translocation was enabled by cyclin D3 (Figure 1B) [108][126][127]. ICP0 has been found to arrest cells in the G2/M phase to promote virus replication by activating the checkpoint kinase 2 (Chk2) [128]. Consistent with these roles of ICP0, it was also found to degrade the centromere proteins CENP-A, CENP-B, and CENP-C, inducing the interphase centromere damage response (iCDR) (Figure 1A) [129][130][131][132]. In addition to this disturbance to the cell cycle, it has been found that ICP0 degrades the DNA-interacting protein TPP1, leading to transcription of telomere repeat-containing RNA activation (TERRA) and increased viral replication [133].
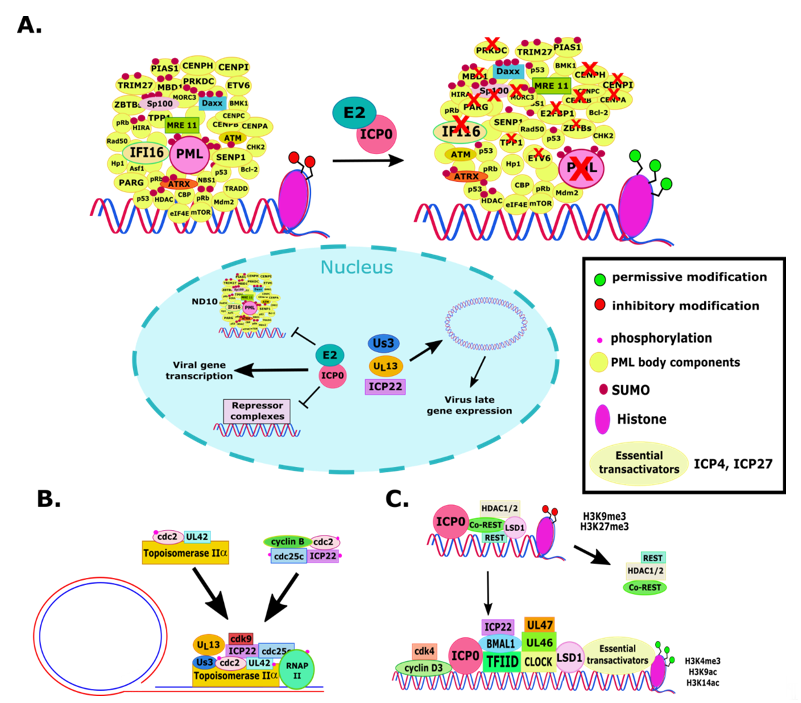
Figure 1. Nuclear functions of non-essential HSV-1 proteins. HSV-1 encodes multiple proteins able to counteract antiviral host responses within the nucleus. (A): ICP0 functions as an E3 ligase ubiquitin ligase that degrades ND10 components that encapsulate the viral genome in the nucleus, including PML, Daxx, SP100, centromeric proteins, and others. The degradation of IFI16 involves multiple factors. These events facilitate initiation of viral gene transcription. (B): The viral protein ICP0 is also known to disrupt repressor complexes that silence the viral genome, as well as recruit factors to enable viral gene transcription. Altogether, ICP0 facilitates permissive histone modifications, while suppressing silencing modifications, to enable for viral gene expression. (C): The viral kinases US3 and UL13, with ICP22, are known to facilitate viral late gene expression, which occurs through the recruitment of host factors, such as Topoisomerase IIα and RNA polymerase II, to the sites of DNA replication in the nucleus. Together, these non-essential viral proteins are important for optimal expression of other viral genes and for viral DNA replication.
As mentioned earlier, there are interwoven relationships between gene silencing and innate immunity and it is not coincidence the ICP0 targets them both. ICP0-null and other ICP0 mutant viruses displayed increased sensitivity to interferon both in vivo and in vitro [32][70][134][135]. As discussed above, ICP0 blocks the nuclear pattern recognition receptors (PRRs) IFI16 and DNA-PKs, which may also impact the cGAS and STING DNA sensing pathway [119][122][123][124][136][137]. Inhibition of STING-dependent immune responses involves ICP0 as ICP0-null virus growth is partially rescued in cells with impaired STING signaling [25][26]. Furthermore, ICP0 was found to reduce the levels of the Toll-like receptor 2 (TLR2) adaptors MyD88 (myeloid differentiation factor 88) and the Mal (MyD88 adaptor-like protein) TIRAP (TIR domain-containing adaptor protein), thus blocking immune responses through this pathway (Figure 2A) [138]. Overall, ICP0 has been proposed to inhibit IRF3 and IRF7-dependent immune responses to sequester these proteins away from host chromatin [139][140][141]. ICP0 was also recently found to have a role in autophagy inhibition through causing the downregulation of p62/SQSTM1 and OPTN autophagy adaptor proteins in a proteasome-dependent and RING finger-independent mechanism (Figure 2C) [142]. It was also demonstrated that the cytoplasmic ICP0 is most likely involved in this function. Another target of ICP0 is the deubiquitylating enzyme USP7 (ubiquitin-specific protease 7) or HAUSP. USP7 appears to bind and stabilize ICP0, but ICP0 degrades USP7 late during infection in a RING finger-dependent manner [58][78][79][143][144]. One reason why the virus could promote degradation of USP7 is because it has a major role in TLR- and TNFa receptor (TNFR)-induced gene expression [145].
Most functions of ICP0 discussed above are performed while in the nucleus. However, ICP0 translocates to the cytoplasm after enabling viral gene expression, where it remains for the reminder of the infection. The cytoplasmic functions of ICP0 remain unexplored. Dr. Roizman’s group first described an interaction of ICP0 with the endocytosis adaptor CIN85 [146]. Dr. Kalamvoki’s group has built upon these findings and reported that ICP0 promotes endocytosis of the viral entry receptor Nectin-1 (Figure 2A) [147]. This is perhaps a mechanism that ensures spread of progeny viruses to uninfected cells. CIN85 forms a complex with the Cbl E3 ligase that is involved in endocytosis of multiple surface components. Thus, ICP0 through CIN85 and Cbl could modulate the surface of infected cells to suppress antiviral responses.
Finally, the role of ICP0 has also been investigated during the latent stage of the virus. ICP0 appears to be important for efficient virus reactivation from latently infected trigeminal ganglia (TG) in mouse ocular infections [67][68][71][148][149][150][151]. ICP0 is also required for VP16-dependent viral reactivation [67][152]. While ICP0 is important for balancing lytic and latent infection, it is still not fully understood what its specific role is in this process.
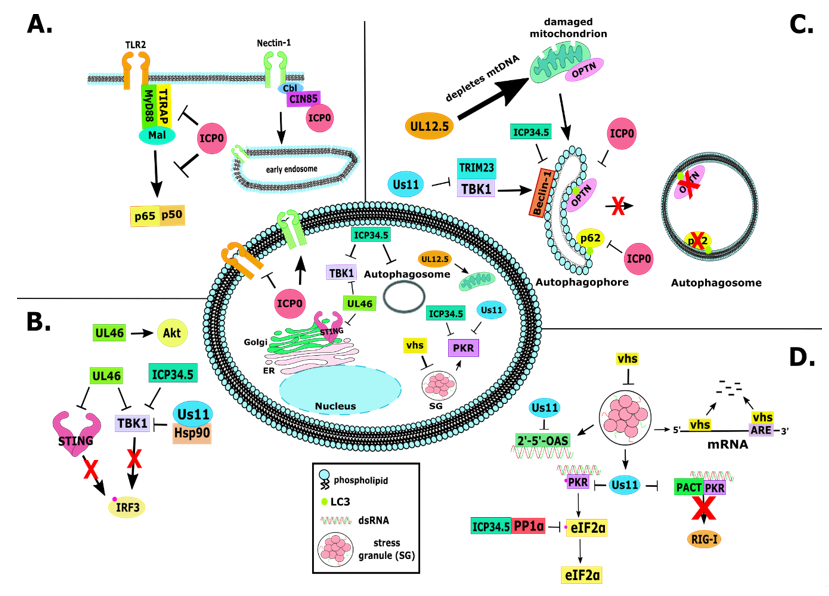
Figure 2. Cytoplasmic functions of some non-essential HSV-1 proteins. (A): ICP0 participates in two major functions in the cytoplasm. First, ICP0 degrades the TLR2 adaptors TIRAP and Mal, thus blocking NF-κB activity. ICP0 also binds to the endocytosis adaptor CIN85 and along with Cbl promotes internalization of the viral entry receptor Nectin-1. This is a mechanism to promote progeny virus spread to uninfected cells. (B): The tegument protein UL46 blocks STING and TBK1, which prevents stimulation of interferon-regulated genes. ICP34.5 and US11 are also involved in blocking TBK1, emphasizing the importance of blocking TBK1 activity during HSV-1 infection. (C): The autophagy pathway is blocked during HSV-1 infection through binding of ICP34.5 to Beclin-1, thus preventing maturation of the autophagophore to an autophagosome. ICP0 has also been found to cause downregulation of p62 and OPTN proteins during infection, which may also serve as another mechanism of blocking selective autophagy. It has also been found that the protein encoded by UL12.5 causes depletion of mtDNA during infection, which causes damage to mitochondria. (D): HSV-1 prevents host translational shutoff from occurring during infection. One mechanism is through ICP34.5 binding to both PP1a and eIF2α, causing dephosphorylation of eIF2α and preventing shutoff of translation. HSV-1 also encodes vhs, which is a viral RNase that degrades AU-rich element (ARE) containing mRNAs. It has also been shown that vhs prevents the formation of cytoplasmic stress granules (SGs) during infection, which contain dsRNA that would otherwise cause PKR activation. HSV-1 also encodes US11, which blocks PKR, thus blocking host translational shutoff and innate immunity activation, as well as blocking PKR and PACT-induced activation of RIG-I during infection.
References
- McGeoch, D.J.; Dolan, A.; Ralph, A.C. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. Virol. 2000, 74, 10401–10406, doi:10.1128/jvi.74.22.10401-10406.2000.
- Underdown, S.J.; Kumar, K.; Houldcroft, C. Network analysis of the hominin origin of Herpes Simplex virus 2 from fossil data. Virus Evol. 2017, 3, doi:10.1093/ve/vex026.
- Knipe, D.M.; Howley, P. Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1.
- Mador, N.; Goldenberg, D.; Cohen, O.; Panet, A.; Steiner, I. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. Virol. 1998, 72, 5067–5075, doi:10.1128/JVI.72.6.5067-5075.1998.
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783, doi:10.1038/nature07103.
- Shen, W.; Sa e Silva, M.; Jaber, T.; Vitvitskaia, O.; Li, S.; Henderson, G.; Jones, C. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. Virol. 2009, 83, 9131–9139, doi:10.1128/JVI.00871-09.
- Croen, K.D.; Ostrove, J.M.; Dragovic, L.J.; Smialek, J.E.; Straus, S.E. Latent herpes simplex virus in human trigeminal ganglia. Detection of an immediate early gene “anti-sense” transcript by in situ hybridization. Engl. J. Med. 1987, 317, 1427–1432, doi:10.1056/NEJM198712033172302.
- Mador, N.; Panet, A.; Latchman, D.; Steiner, I. Expression and splicing of the latency-associated transcripts of herpes simplex virus type 1 in neuronal and non-neuronal cell lines. Biochem. 1995, 117, 1288–1297, doi:10.1093/oxfordjournals.jbchem.a124857.
- Garber, D.A.; Schaffer, P.A.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. Virol. 1997, 71, 5885–5893, doi:10.1128/JVI.71.8.5885-5893.1997.
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. Virol. 1997, 71, 5878–5884, doi:10.1128/JVI.71.8.5878-5884.1997.
- Wagner, E.K.; Bloom, D.C. Experimental investigation of herpes simplex virus latency. Microbiol. Rev. 1997, 10, 419–443, doi:10.1128/CMR.10.3.419.
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. Virol. 2002, 76, 717–729, doi:10.1128/JVI.76.2.717-729.2002.
- Henderson, G.; Peng, W.; Jin, L.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. Neurovirol. 2002, 8, 103–111, doi:10.1080/13550280290101085.
- Inman, M.; Perng, G.-C.; Henderson, G.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. Virol. 2001, 75, 3636–3646, doi:10.1128/JVI.75.8.3636-3646.2001.
- Jin, L.; Peng, W.; Perng, G.-C.; Brick, D.J.; Nesburn, A.B.; Jones, C.; Wechsler, S.L. Identification of herpes simplex virus type 1 latency-associated transcript sequences that both inhibit apoptosis and enhance the spontaneous reactivation phenotype. Virol. 2003, 77, 6556–6561, doi:10.1128/JVI.77.11.6556-6561.2003.
- Perng, G.C.; Ghiasi, H.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. Virol. 1996, 70, 976–984.
- Peng, W.; Henderson, G.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. The gene that encodes the herpes simplex virus type 1 latency-associated transcript influences the accumulation of transcripts (Bcl-xL and Bcl-xS) that encode apoptotic regulatory proteins. Virol. 2003, 77, 10714–10718, doi:10.1128/JVI.77.19.10714-10718.2003.
- Perng, G.-C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503, doi:10.1126/science.287.5457.1500.
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. Virol. 1974, 14, 8–19, doi:10.1128/JVI.14.1.8-19.1974.
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis: Sequential transition of polypeptide synthesis requires functional viral polypeptides. Natl. Acad. Sci. USA 1975, 72, 1276–1280, doi:10.1073/pnas.72.4.1276.
- Longnecker, R.; Roizman, B. Clustering of genes dispensable for growth in culture in the S component of the HSV-1 genome. Science 1987, 236, 573–576, doi:10.1126/science.3033823.
- McGeoch, D.J.; Dolan, A.; Frame, M.C. DNA sequence of the region in the genome of herpes simplex virus type 1 containing the exonuclease gene and neighbouring genes. Nucleic Acids Res. 1986, 14, 3435–3448, doi:10.1093/nar/14.8.3435.
- Toh, Y.; Tanaka, S.; Liu, Y.; Hidaka, Y.; Mori, R. Molecular characterization of naturally occuring glycoprotein C-negative herpes simplex virus type 1. Virol. 1993, 129, 119–130.
- Hidaka, Y.; Sakuma, S.; Kumano, Y.; Minagawa, H.; Mori, R. Characterization of glycoprotein C-negative mutants of herpes simplex virus type 1 isolated from a patient with keratitis. Virol. 1990, 113, 195–207, doi:10.1007/BF01316673.
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Natl. Acad. Sci. USA 2014, 111, E611-7, doi:10.1073/pnas.1323414111.
- Deschamps, T.; Kalamvoki, M. Impaired STING pathway in human osteosarcoma U2OS cells contributes to the growth of ICP0-null mutant herpes simplex virus. Virol. 2017, 91, doi:10.1128/JVI.00006-17.
- Ackermann, M.; Braun, D.K.; Pereira, L.; Roizman, B. Characterization of herpes simplex virus 1 alpha proteins 0, 4, and 27 with monoclonal antibodies. Virol. 1984, 52, 108–118.
- Stow, N.D.; Stow, E.C. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. Gen. Virol. 1986, 67, 2571–2585, doi:10.1099/0022-1317-67-12-2571.
- Sacks, W.R.; Schaffer, P.A. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. Virol. 1987, 61, 829–839, doi:10.1128/JVI.61.3.829-839.1987.
- Yao, F.; Schaffer, P.A. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. Virol. 1995, 69, 6249–6258, doi:10.1128/JVI.69.10.6249-6258.1995.
- Chen, J.; Silverstein, S. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. Virol. 1992, 66, 2916–2927, doi:10.1128/JVI.66.5.2916-2927.1992.
- Everett, R.D.; Boutell, C.; Orr, A. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. Virol. 2004, 78, 1763–1774, doi:10.1128/jvi.78.4.1763-1774.2004.
- Kalamvoki, M.; Roizman, B. Role of herpes simplex virus ICP0 in the transactivation of genes introduced by infection or transfection: A reappraisal. Virol. 2010, 84, 4222–4228, doi:10.1128/JVI.02585-09.
- Alandijany, T.; Roberts, A.P.E.; Conn, K.L.; Loney, C.; McFarlane, S.; Orr, A.; Boutell, C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog. 2018, 14, e1006927, doi:10.1371/journal.ppat.1006769.
- Sandri-Goldin, R.M.; Sekulovich, R.E.; Leary, K. The alpha protein ICP0 does not appear to play a major role in the regulation of herpes simplex virus gene expression during infection in tissue culture. Nucleic Acids Res. 1987, 15, 905–919, doi:10.1093/nar/15.3.905.
- Moriuchi, H.; Moriuchi, M.; Straus, S.E.; Cohen, J.I. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. Virol. 1993, 67, 4290–4295, doi:10.1128/JVI.67.7.4290-4295.1993.
- Everett, R.; Orr, A.; Elliott, M. The equine herpesvirus 1 gene 63 RING finger protein partially complements Vmw110, its herpes simplex virus type 1 counterpart. Gen. Virol. 1995, 76, 2369–2374, doi:10.1099/0022-1317-76-9-2369.
- Everett, R.D.; Boutell, C.; McNair, C.; Grant, L.; Orr, A. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. Virol. 2010, 84, 3476–3487, doi:10.1128/JVI.02544-09.
- Quinlan, M.P.; Knipe, D.M. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Cell. Biol. 1985, 5, 957–963, doi:10.1128/MCB.5.5.957.
- Knipe, D.M.; Smith, J.L. A mutant herpesvirus protein leads to a block in nuclear localization of other viral proteins. Cell. Biol. 1986, 6, 2371–2381, doi:10.1128/mcb.6.7.2371.
- Gelman, I.H.; Silverstein, S. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Natl. Acad. Sci. USA 1985, 82, 5265–5269, doi:10.1073/pnas.82.16.5265.
- Mosca, J.D.; Bednarik, D.P.; Raj, N.B.; Rosen, C.A.; Sodroski, J.G.; Haseltine, W.A.; Hayward, G.S.; Pitha, P.M. Activation of human immunodeficiency virus by herpesvirus infection: Identification of a region within the long terminal repeat that responds to a trans-acting factor encoded by herpes simplex virus 1. Natl. Acad. Sci. USA 1987, 84, 7408–7412, doi:10.1073/pnas.84.21.7408.
- Ostrove, J.M.; Leonard, J.; Weck, K.E.; Rabson, A.B.; Gendelman, H.E. Activation of the human immunodeficiency virus by herpes simplex virus type 1. Virol. 1987, 61, 3726–3732, doi:10.1128/JVI.61.12.3726-3732.1987.
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. Virol. 1988, 62, 196–205, doi:10.1128/JVI.62.1.196-205.1988.
- McCusker, C.T.; Bacchetti, S. The responsiveness of human papillomavirus upstream regulatory regions to herpes simplex virus immediate early proteins. Virus Res. 1988, 11, 199–207, doi:10.1016/0168-1702(88)90083-4.
- Gius, D.; Laimins, L.A. Activation of human papillomavirus type 18 gene expression by herpes simplex virus type 1 viral transactivators and a phorbol ester. Virol. 1989, 63, 555–563, doi:10.1128/JVI.63.2.555-563.1989.
- Margolis, D.M.; Rabson, A.B.; Straus, S.E.; Ostrove, J.M. Transactivation of the HIV-1 LTR by HSV-1 immediate-early genes. Virology 1992, 186, 788–791, doi:10.1016/0042-6822(92)90048-t.
- Nabel, G.J.; Rice, S.A.; Knipe, D.M.; Baltimore, D. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science 1988, 239, 1299–1302, doi:10.1126/science.2830675.
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077, doi:10.1371/journal.ppat.1004077.
- O’Hare, P.; Hayward, G.S. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. Virol. 1985, 56, 723–733, doi:10.1128/JVI.56.3.723-733.1985.
- Cai, W.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. Virol. 1992, 66, 2904–2915, doi:10.1128/JVI.66.5.2904-2915.1992.
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. Virol. 2002, 76, 841–850, doi:10.1128/jvi.76.2.841-850.2002.
- Everett, R.D.; Barlow, P.; Milner, A.; Luisi, B.; Orr, A.; Hope, G.; Lyon, D. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. Mol. Biol. 1993, 234, 1038–1047, doi:10.1006/jmbi.1993.1657.
- Grant, K.; Grant, L.; Tong, L.; Boutell, C. Depletion of intracellular zinc inhibits the ubiquitin ligase activity of viral regulatory protein ICP0 and restricts herpes simplex virus 1 replication in cell culture. Virol. 2012, 86, 4029–4033, doi:10.1128/JVI.06962-11.
- Lium, E.K.; Silverstein, S. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. Virol. 1997, 71, 8602–8614, doi:10.1128/JVI.71.11.8602-8614.1997.
- Boutell, C.; Orr, A.; Everett, R.D. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. Virol. 2003, 77, 8686–8694, doi:10.1128/jvi.77.16.8686-8694.2003.
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. Biol. Chem. 2003, 278, 36596–36602, doi:10.1074/jbc.M300776200.
- Boutell, C.; Canning, M.; Orr, A.; Everett, R.D. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. Virol. 2005, 79, 12342–12354, doi:10.1128/JVI.79.19.12342-12354.2005.
- Gu, H.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Natl. Acad. Sci. USA 2003, 100, 8963–8968, doi:10.1073/pnas.1533420100.
- Vanni, E.; Gatherer, D.; Tong, L.; Everett, R.D.; Boutell, C. Functional characterization of residues required for the herpes simplex virus 1 E3 ubiquitin ligase ICP0 to interact with the cellular E2 ubiquitin-conjugating enzyme UBE2D1 (UbcH5a). Virol. 2012, 86, 6323–6333, doi:10.1128/JVI.07210-11.
- Maul, G.G.; Guldner, H.H.; Spivack, J.G. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). Gen. Virol. 1993, 74, 2679–2690, doi:10.1099/0022-1317-74-12-2679.
- Mullen, M.A.; Ciufo, D.M.; Hayward, G.S. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. Virol. 1994, 68, 3250–3266, doi:10.1128/JVI.68.5.3250-3266.1994.
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069.
- Everett, R.D.; Murray, J.; Orr, A.; Preston, C.M. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. Virol. 2007, 81, 10991–11004, doi:10.1128/JVI.00705-07.
- Cohen, C.; Corpet, A.; Roubille, S.; Maroui, M.A.; Poccardi, N.; Rousseau, A.; Kleijwegt, C.; Binda, O.; Texier, P.; Sawtell, N.; et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog. 2018, 14, e1007313, doi:10.1371/journal.ppat.1007313.
- Maroui, M.A.; Callé, A.; Cohen, C.; Streichenberger, N.; Texier, P.; Takissian, J.; Rousseau, A.; Poccardi, N.; Welsch, J.; Corpet, A.; et al. Latency entry of herpes simplex Virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog. 2016, 12, e1005834, doi:10.1371/journal.ppat.1005834.
- Thompson, R.L.; Sawtell, N.M. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. Virol. 2006, 80, 10919–10930, doi:10.1128/JVI.01253-06.
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. Virol. 1993, 67, 7501–7512, doi:10.1128/JVI.67.12.7501-7512.1993.
- Roizman, B. The checkpoints of viral gene expression in productive and latent infection: The role of the HDAC/CoREST/LSD1/REST repressor complex. Virol. 2011, 85, 7474–7482, doi:10.1128/JVI.00180-11.
- Härle, P.; Sainz, B.J.; Carr, D.J.J.; Halford, W.P. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 2002, 293, 295–304, doi:10.1006/viro.2001.1280.
- Halford, W.P.; Schaffer, P.A. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. Virol. 2001, 75, 3240–3249, doi:10.1128/JVI.75.7.3240-3249.2001.
- Deschamps, T.; Waisner, H.; Dogrammatzis, C.; Roy, A.; Chacko, S.; Perera, C.; Prisinzano, T.E.; Kalamvoki, M. Discovery of small-molecule inhibitors targeting the E3 ubiquitin ligase activity of the herpes simplex virus 1 ICP0 protein using an in vitro high-throughput screening assay. Virol. 2019, 93, doi:10.1128/JVI.00619-19.
- Hagglund, R.; Roizman, B. Herpes simplex virus 1 mutant in which the ICP0 HUL-1 E3 ubiquitin ligase site is disrupted stabilizes cdc34 but degrades D-type cyclins and exhibits diminished neurotoxicity. Virol. 2003, 77, 13194–13202, doi:10.1128/jvi.77.24.13194-13202.2003.
- Gu, H.; Poon, A.P.; Roizman, B. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Natl. Acad. Sci. U.S.A 2009, 106, 19132–19137, doi:10.1073/pnas.0910920106.
- Boutell, C.; Cuchet-Lourenço, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245, doi:10.1371/journal.ppat.1002245.
- Cuchet-Lourenço, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. Virol. 2012, 86, 11209–11222, doi:10.1128/JVI.01145-12.
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 proteome during HSV-1 infection. PLoS Pathog. 2015, 11, e1005059, doi:10.1371/journal.ppat.1005059.
- Everett, R.D.; Boutell, C.; Pheasant, K.; Cuchet-Lourenço, D.; Orr, A. Sequences related to SUMO interaction motifs in herpes simplex virus 1 protein ICP0 act cooperatively to stimulate virus infection. Virol. 2014, 88, 2763–2774, doi:10.1128/JVI.03417-13.
- Everett, R.D.; Parsy, M.-L.; Orr, A. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. Virol. 2009, 83, 4963–4977, doi:10.1128/JVI.02593-08.
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. Virol. 1998, 72, 6581–6591, doi:10.1128/JVI.72.8.6581-6591.1998.
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941, doi:10.1038/sj.onc.1202366.
- Sha, Z.; Blyszcz, T.; González-Prieto, R.; Vertegaal, A.C.O.; Goldberg, A.L. Inhibiting ubiquitination causes an accumulation of SUMOylated newly synthesized nuclear proteins at PML bodies. Biol. Chem. 2019, 294, 15218–15234, doi:10.1074/jbc.RA119.009147.
- Everett, R.D.; Murray, J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. Virol. 2005, 79, 5078–5089, doi:10.1128/JVI.79.8.5078-5089.2005.
- Maroui, M.A.; Maarifi, G.; McManus, F.P.; Lamoliatte, F.; Thibault, P.; Chelbi-Alix, M.K. Promyelocytic Leukemia Protein (PML) requirement for interferon-induced global cellular SUMOylation. Cell. Proteomics 2018, 17, 1196–1208, doi:10.1074/mcp.RA117.000447.
- Sloan, E.; Orr, A.; Everett, R.D. MORC3, a Component of PML nuclear bodies, has a role in restricting herpes simplex virus 1 and human cytomegalovirus. Virol. 2016, 90, 8621–8633, doi:10.1128/JVI.00621-16.
- Conn, K.L.; Wasson, P.; McFarlane, S.; Tong, L.; Brown, J.R.; Grant, K.G.; Domingues, P.; Boutell, C. Novel role for protein inhibitor of activated STAT 4 (PIAS4) in the restriction of herpes simplex virus 1 by the cellular intrinsic antiviral immune response. Virol. 2016, 90, 4807–4826, doi:10.1128/JVI.03055-15.
- Brown, J.R.; Conn, K.L.; Wasson, P.; Charman, M.; Tong, L.; Grant, K.; McFarlane, S.; Boutell, C. SUMO ligase protein inhibitor of activated STAT1 (PIAS1) is a constituent promyelocytic leukemia nuclear body protein that contributes to the intrinsic antiviral immune response to herpes simplex virus 1. Virol. 2016, 90, 5939–5952, doi:10.1128/JVI.00426-16.
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. Virol. 1996, 70, 7471–7477, doi:10.1128/JVI.70.11.7471-7477.1996.
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. Virol. 1999, 73, 650–657, doi:10.1128/JVI.73.1.650-657.1999.
- Jackson, S.A.; DeLuca, N.A. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Natl. Acad. Sci. USA 2003, 100, 7871–7876, doi:10.1073/pnas.1230643100.
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–55, doi:10.1038/emboj.2009.400.
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084, doi:10.1371/journal.ppat.1002084.
- Chaurushiya, M.S.; Lilley, C.E.; Aslanian, A.; Meisenhelder, J.; Scott, D.C.; Landry, S.; Ticau, S.; Boutell, C.; Yates, J.R., III; Schulman, B.A.; et al. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Cell 2012, 46, 79–90, doi:10.1016/j.molcel.2012.02.004.
- Nagel, C.-H.; Albrecht, N.; Milovic-Holm, K.; Mariyanna, L.; Keyser, B.; Abel, B.; Weseloh, B.; Hofmann, T.G.; Eibl, M.M.; Hauber, J. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. Virol. 2011, 85, 7644–7657, doi:10.1128/JVI.02207-10.
- Conwell, S.E.; White, A.E.; Harper, J.W.; Knipe, D.M. Identification of TRIM27 as a novel degradation target of herpes simplex virus 1 ICP0. Virol. 2015, 89, 220–229, doi:10.1128/JVI.02635-14.
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. Virol. 1989, 63, 943–947, doi:10.1128/JVI.63.2.943-947.1989.
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. Virol. 2004, 78, 9689–9696, doi:10.1128/JVI.78.18.9689-9696.2004.
- Kent, J.R.; Zeng, P.-Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. Virol. 2004, 78, 10178–10186, doi:10.1128/JVI.78.18.10178-10186.2004.
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.M.; Zeng, P.-Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. Virol. 2006, 80, 5740–5746, doi:10.1128/JVI.00169-06.
- Cabral, J.M.; Oh, H.S.; Knipe, D.M. ATRX promotes maintenance of herpes simplex virus heterochromatin during chromatin stress. eLife 2018, 7, doi:10.7554/eLife.40228.
- Lee, J.S.; Raja, P.; Knipe, D.M. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio 2016, 7, doi:10.1128/mBio.02007-15.
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. Virol. 2004, 78, 6744–6757, doi:10.1128/JVI.78.13.6744-6757.2004.
- Poon, A.P.W.; Gu, H.; Roizman, B. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Natl. Acad. Sci. USA 2006, 103, 9993–9998, doi:10.1073/pnas.0604142103.
- Hobbs, W.E., II; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. Virol. 1999, 73, 8245–8255, doi:10.1128/JVI.73.10.8245-8255.1999.
- Poon, A.P.W.; Liang, Y.; Roizman, B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. Virol. 2003, 77, 12671–12678, doi:10.1128/jvi.77.23.12671-12678.2003.
- Kalamvoki, M.; Roizman, B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Natl. Acad. Sci. USA 2010, 107, 17721–17726, doi:10.1073/pnas.1012991107.
- Kalamvoki, M.; Roizman, B. The Histone Acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. Virol. 2011, 85, 9472–9477, doi:10.1128/JVI.00876-11.
- Kalamvoki, M.; Roizman, B. Interwoven roles of cyclin D3 and cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. Virol. 2010, 84, 9709–9717, doi:10.1128/JVI.01050-10.
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. Virol. 2008, 82, 12030–12038, doi:10.1128/JVI.01575-08.
- Kutluay, S.B.; Triezenberg, S.J. Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. Virol. 2009, 83, 5835–5845, doi:10.1128/JVI.00219-09.
- Placek, B.J.; Huang, J.; Kent, J.R.; Dorsey, J.; Rice, L.; Fraser, N.W.; Berger, S.L. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. Virol. 2009, 83, 1416–1421, doi:10.1128/JVI.01276-08.
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. Virol. 2009, 83, 4376–4385, doi:10.1128/JVI.02515-08.
- Gu, H.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Natl. Acad. Sci. USA 2005, 102, 7571–7576, doi:10.1073/pnas.0502658102.
- Gu, H.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Natl. Acad. Sci. USA 2007, 104, 17134–17139, doi:10.1073/pnas.0707266104.
- Kalamvoki, M.; Roizman, B. Nuclear retention of ICP0 in cells exposed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Natl. Acad. Sci. USA 2008, 105, 20488–20493, doi:10.1073/pnas.0810879105.
- Merkl, P.E.; Orzalli, M.H.; Knipe, D.M. Mechanisms of host IFI16, PML, and Daxx protein restriction of herpes simplex virus 1 replication. Virol. 2018, 92, doi:10.1128/JVI.00057-18.
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. Virol. 2010, 84, 4026–4040, doi:10.1128/JVI.02597-09.
- Everett, R.D. Dynamic response of IFI16 and promyelocytic leukemia nuclear body components to herpes simplex virus 1 infection. Virol. 2016, 90, 167–179, doi:10.1128/JVI.02249-15.
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 2016, 7, doi:10.1128/mBio.01553-16.
- McFarlane, S.; Orr, A.; Roberts, A.P.E.; Conn, K.L.; Iliev, V.; Loney, C.; Filipe, A.d.S.; Smollett, K.; Gu, Q.; Robertson, N.; et al. The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV-1 infection. PLoS Pathog. 2019, 15, e1007667, doi:10.1371/journal.ppat.1007667.
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. Virol. 2012, 86, 10093–10102, doi:10.1128/JVI.00930-12.
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Natl. Acad. Sci. USA 2012, 109, doi:10.1073/pnas.1211302109.
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. Virol. 2016, 90, 8351–8359, doi:10.1128/JVI.00939-16.
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. Virol. 2013, 87, 13422–13432, doi:10.1128/JVI.02474-13.
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Cell. Proteomics 2015, 14, 2341–2356, doi:10.1074/mcp.M114.047068.
- Kawaguchi, Y.; van Sant, C.; Roizman, B. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. Virol. 1997, 71, 7328–7336, doi:10.1128/JVI.71.10.7328-7336.1997.
- Kalamvoki, M.; Roizman, B. ICP0 enables and monitors the function of D cyclins in herpes simplex virus 1 infected cells. Natl. Acad. Sci. USA 2009, 106, 14576–14580, doi:10.1073/pnas.0906905106.
- Li, H.; Baskaran, R.; Krisky, D.M.; Bein, K.; Grandi, P.; Cohen, J.B.; Glorioso, J.C. Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 2008, 375, 13–23, doi:10.1016/j.virol.2008.01.038.
- Lomonte, P.; Sullivan, K.F.; Everett, R.D. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. Biol. Chem. 2001, 276, 5829–5835, doi:10.1074/jbc.M008547200.
- Lomonte, P.; Morency, E. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 2007, 581, 658–662, doi:10.1016/j.febslet.2007.01.027.
- Everett, R.D.; Earnshaw, W.C.; Findlay, J.; Lomonte, P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 1999, 18, 1526–1538, doi:10.1093/emboj/18.6.1526.
- Gross, S.; Catez, F.; Masumoto, H.; Lomonte, P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS ONE 2012, 7, e44227, doi:10.1371/journal.pone.0044227.
- Deng, Z.; Kim, E.T.; Vladimirova, O.; Dheekollu, J.; Wang, Z.; Newhart, A.; Liu, D.; Myers, J.L.; Hensley, S.E.; Moffat, J.; et al. HSV-1 remodels host telomeres to facilitate viral replication. Cell Rep. 2014, 9, 2263–2278, doi:10.1016/j.celrep.2014.11.019.
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. Virol. 2000, 74, 2052–2056, doi:10.1128/jvi.74.4.2052-2056.2000.
- Leib, D.A.; Harrison, T.E.; Laslo, K.M.; Machalek, M.A.; Moorman, N.J.; Virgin, H.W. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. Exp. Med. 1999, 189, 663–672, doi:10.1084/jem.189.4.663.
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M.J.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Immunol. 2020, 5, doi:10.1126/sciimmunol.aba4219.
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Natl. Acad. Sci. USA 2015, 112, 1773–1781, doi:10.1073/pnas.1424637112.
- Van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. Virol. 2010, 84, 10802–10811, doi:10.1128/JVI.00063-10.
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. Virol. 2004, 78, 1675–1684, doi:10.1128/JVI.78.4.1675-1684.2004.
- Melroe, G.T.; DeLuca, N.A.; Knipe, D.M. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. Virol. 2004, 78, 8411–8420, doi:10.1128/JVI.78.16.8411-8420.2004.
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 2007, 360, 305–321, doi:10.1016/j.virol.2006.10.028.
- Waisner, H.; Kalamvoki, M. The ICP0 protein of herpes simplex virus 1 (HSV-1) downregulates major autophagy adaptor proteins sequestosome 1 and optineurin during the early stages of HSV-1 infection. Virol. 2019, 93, doi:10.1128/JVI.01258-19.
- Kalamvoki, M.; Gu, H.; Roizman, B. Overexpression of the ubiquitin-specific protease 7 resulting from transfection or mutations in the ICP0 binding site accelerates rather than depresses herpes simplex virus 1 gene expression. Virol. 2012, 86, 12871–12878, doi:10.1128/JVI.01981-12.
- Mostafa, H.H.; Thompson, T.W.; Davido, D.J. N-terminal phosphorylation sites of herpes simplex virus 1 ICP0 differentially regulate its activities and enhance viral replication. Virol. 2013, 87, 2109–2119, doi:10.1128/JVI.02588-12.
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-κB by ubiquitin-specific protease-7 promotes transcription. Natl. Acad. Sci. USA 2013, 110, 618–623, doi:10.1073/pnas.1208446110.
- Liang, Y.; Kurakin, A.; Roizman, B. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Natl. Acad. Sci. USA 2005, 102, 5838–5843, doi:10.1073/pnas.0501253102.
- Deschamps, T.; Dogrammatzis, C.; Mullick, R.; Kalamvoki, M. Cbl E3 ligase mediates the removal of nectin-1 from the surface of herpes simplex virus 1-infected cells. Virol. 2017, 91, doi:10.1128/jvi.00393-17.
- Leib, D.A.; Coen, D.M.; Bogard, C.L.; Hicks, K.A.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; Schaffer, P.A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. Virol. 1989, 63, 759–768, doi:10.1128/JVI.63.2.759-768.1989.
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. Virol. 2001, 75, 6143–6153, doi:10.1128/JVI.75.13.6143-6153.2001.
- Harris, R.A.; Everett, R.D.; Zhu, X.X.; Silverstein, S.; Preston, C.M. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. Virol. 1989, 63, 3513–3515, doi:10.1128/JVI.63.8.3513-3515.1989.
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 2015, 18, 649–658, doi:10.1016/j.chom.2015.11.007.
- Thompson, R.L.; Preston, C.M.; Sawtell, N.M. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009, 5, e1000352, doi:10.1371/journal.ppat.1000352.

