 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antoine Dufour | + 1303 word(s) | 1303 | 2021-01-06 03:42:08 | | | |
| 2 | Camila Xu | Meta information modification | 1303 | 2021-01-11 04:23:19 | | | | |
| 3 | Camila Xu | Meta information modification | 1303 | 2021-03-18 03:57:34 | | |
Video Upload Options
Matrix metalloproteinases (MMPs) are zinc-dependent proteases that have been extensively studied in the context of extracellular matrix (ECM) breakdown and remodelling.
1. Introduction
Increasingly, non-ECM substrates are being investigated for MMPs as ECM substrates only account for approximately 30% of all known MMP substrates [1][2]. The dysregulation of MMPs, their substrates, and the tissue inhibitor of metalloproteinases (TIMPs) often results in the progression of numerous diseases [3][2][4]. Various MMPs have been implicated in multiple cancers including pancreas, brain, lung, prostate, breast, skin and gastrointestinal tract [2][5]. MMP12 has been studied in chronic obstructive pulmonary disease (COPD) and the minor allele of a single nucleotide polymorphism in MMP12 (rs2276109) was associated with a beneficial effect on lung function in smokers and children with asthma [6][7]. Multiple MMPs have been investigated in rheumatoid arthritis and osteoarthritis yet the precise functions of individual MMP remains to be better characterized (reviewed in [8]). MMPs have also been studied in context of periodontal diseases [9][10]. It is not surprising that MMP inhibitors were tested in clinical trials. However, to date, the only MMP inhibitor that is currently approved is Periostat® (doxycycline hyclate), which is used for treating periodontitis (Figure 1a). Despite their biological roles in multiple cancers, in addition to inflammatory and autoimmune diseases, most MMP inhibitors failed due to a combination of factors including poor study design, a lack of understanding of biological roles of MMPs and the substrates they cleave, and the lack of specific inhibitors [3][2][5][11]. The structures and amino acid sequence of the catalytic domain of the 23 MMPs are highly conserved, which initially resulted in the design of broad spectrum MMP inhibitors. MMPs, however, have both detrimental and protective functions, limiting the use of these broad-spectrum inhibitors and increasing the complexity of developing MMP inhibitors to treat human diseases.
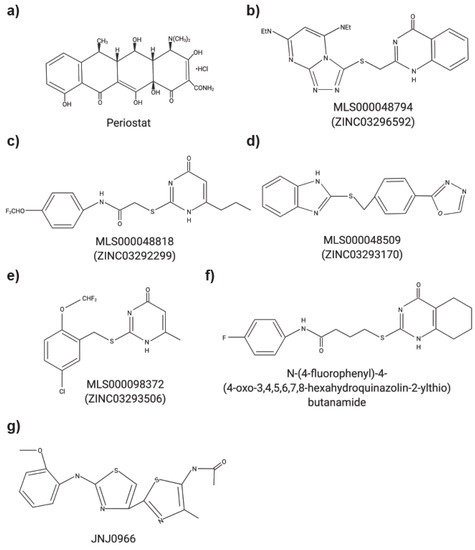
Figure 1. Chemical structures of small molecule MMP inhibitors. (a) Periostat®: doxycycline hyclate. (b) MLS000048794: 2-[[3,5bis(ethylamino)-2,4,6,8,9-pentazabicyclo[4 .3.0]nona-2,4,7,9-tetraen-7yl] sulfanyl-methyl]-1H-quinazolin4-one). (c) MLS000048818: N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide. (d) MLS000048509: 2-[[4-(1,3,4-oxadiazol-2-yl)phenyl] methylsulfanyl]-1Hbenzoimidazole). (e) MLS000098372: 2-[[5-chloro-2 (difluoromethoxy)phenyl]methylsulfanyl]-6-methyl-1H-pyrimidin-4-one). (f) N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide. (g) JNJ0966: N-{2-[(2-methoxyphenyl)amino]-4′-methyl-4,5′-bi-1,3-thiazol-2′-yl}acetamide.
2. Regulation of MMP Activity
The catalytic activity of MMPs is tightly regulated by endogenous TIMPs [12]. TIMPs are secreted proteins that inhibit metalloproteinases [13] through the formation of 1:1 stoichiometric complexes [12]. The C-terminus of TIMPs interacts with the hemopexin like domain, found in all MMPs except MMP7 and MMP26, whereas the N-terminus interacts with the zinc ion within the catalytic domains of MMPs [14]. When an imbalance between MMPs and TIMPs occurs, it often results in inflammation and immune responses, as seen in many inflammatory diseases and cancers [15]. Therefore, the reestablishment of MMP-TIMP homeostasis is of pharmacological value and supports the need for the development of effective MMP inhibitors. Moreover, a better understanding of the biological functions of TIMPs is also needed to clarify their roles in human pathologies.
3. Non-Proteolytic Functions of MMPs
As demonstrated in previous clinical trials, broad spectrum targeting of the catalytic domain of MMPs is challenging. Thus, alternative methods for the inhibition of MMP functions have been investigated such as targeting exosites and ectosites. Not only would this potentially enable greater specificity between MMPs, but some exosites may have unique functions distinct from proteolysis. One example is the hemopexin (PEX) domain that contributes to protein-protein interactions and can initiate cell signalling and increased cell migration [16][17][18]. Since the amino acid sequences of the PEX domain across MMPs is more divergent and less conserved than the catalytic domain, the PEX domain is a potential site to target with inhibitors to increase selectivity. Interestingly, MMP7 and MMP26 do not contain a PEX domain, therefore not all MMPs require this domain. The MMP1 PEX domain is essential for binding to collagen and in the modulation of the triple helical structure of the substrate to allow access to the catalytic cleft [19]. Both the catalytic and PEX domain of MMP1 are necessary for the cooperative binding of triple helix collagen, demonstrating the importance of the PEX domain in substrate binding. Additionally, the conserved collagen residue P10 interacts with MMP1 via a hydrophobic pocket or exosite composed of Phe301, Ile271, and Arg27 within the PEX domain [19]. Further, when double mutants of Ile271Ala/Arg272Ala were generated, the collagenolytic function was significantly reduced. Thus, the inhibition of this hydrophobic pocket could potentially be a therapeutic approach to regulate MMP1 activity as it is important in not only the binding of triple helix collagen but in the processing of collagen [19]. The PEX domain of MMP12 plays a critical role in clearance of various bacteria such as Staphylococcus aureus, Klebsiella pneumoniae, Escherichia coli, and Salmonella enteriditis in the phagolysosome [18]. In Mmp12−/− mice, there was an increase in mortality at lower titer concentration when infected with S. aureus as compared with wild type mice [18]. Anti-bacterial properties of MMP12 were determined to be the result of disruption of the bacterial outer membrane by amino acids 344-363 in blade II of the PEX domain [18]. Conversely, the catalytic domain of MMP12 may contribute to the cleavage of bacterial toxins but did not demonstrate antibacterial properties against S. aureus α-toxins [18]. Therefore, a better characterization of the PEX domains of MMPs may reveal new exciting functions in other MMPs.
The PEX domain of MMPs is also implicated in homo-/hetero-dimerization and can form multimers [20]. The propeller structure of the PEX domain includes 4 blades composed of two alpha-helices and four beta strands [21]. In MMP9, a mutation in blade IV of the PEX domain resulted in a loss of homodimer formation [16]. Mutations in blade I of the MMP9 PEX domain resulted in a loss of interactions with the cell surface CD44 [16]. This interaction between the outer blade I of the MMP9 PEX domain and CD44 was shown to increase cell migration via the activation of epidermal growth factor receptor (EGFR) and downstream kinase signaling [16]. Peptides generated to mimic the outer beta strand of blade I or IV resulted in decreased levels of MMP9 dimers and also a reduction cell migration [16]. MMP9 can also increase angiogenesis [22]. Using an allosteric inhibitor to the PEX domain, Hariono et al. [22] demonstrated that inhibition of ECM proteolysis, which decreases the release of vascular endothelial growth factor (VEGF) from within the ECM, significantly reduces the binding of VEGF to its membrane receptor, and subsequently decreases angiogenesis.
The catalytic domain of membrane type 1-matrix metalloproteinase/MT1-MMP (MMP14) has been implicated in pro-tumorigenic functions by processing type I collagen, in addition to increasing cell migration, angiogenesis, and cell invasion [23][24][25]. The PEX domain of MT1-MMP also forms hetero- (with CD44) and homo-dimers via blades I and IV of the PEX domain, respectively [25]. Synthetic peptides mimicking the outermost strand motifs within the PEX domain (blades I and IV) of MT1-MMP were shown to specifically inhibit MT1-MMP-enhanced cell migration, although the ability to directly prevent MT1-MMP proteolytic activity was not shown [25]. The PEX domain contributes to the tumor promoting nature of MT1-MMP as tumour volume was significantly larger in cancer cells containing the PEX domain compared to those without [24]. MT1-MMP also contains transmembrane and cytoplasmic tail domains that have been shown to have distinct functions from the catalytic domain and could be targeted with inhibitors to interfere with the biological functions of MT1-MMP. Targeting the PEX domain of MMPs could provide non-competitive inhibition as compared with active site inhibition with broad-spectrum compounds [26]. Each MMP is likely to have unique exosites or “hotspots” that may be targeted individually due to divergence of their amino acid sequences, chemical potential and geometry [27]. However, the binding affinity of most exosites for substrate is typically low (10−6–10−7 M) making it potentially challenging to design an effective drug against that site [28][29].
References
- Dufour, A.; Overall, C.M. Subtracting Matrix Out of the Equation: New Key Roles of Matrix Metalloproteinases in Innate Immunity and Disease. In Matrix Metalloproteinase Biology, 1st ed.; Sagi, I., Gaffney, J., Eds.; John Wiley & Sons, Inc.: Franklin Township, NJ, USA, 2015; pp. 131–152.
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242.
- Young, D.; Das, N.; Anowai, A.; Dufor, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847.
- Mainoli, B.; Hirota, S.; Edgington-Mitchell, L.E. Proteomics and Imaging in Crohn’s Disease: TAILS of Unlikely Allies. Trends Pharmacol. Sci. 2020, 41, 74–84.
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239.
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melén, E.; Söderhäll, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608.
- Mallia-Milanes, B.; Dufour, A.; Philp, C.; Solis, N.; Klein, T.; Fischer, M.; Bolton, C.E.; Shapiro, S.; Overall, C.M.; Johnson, S.R. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L1003–L1014.
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135.
- Checchi, V.; Maravic, T.; Bellini, P.; Generali, L.; Consolo, U.; Breschi, L.; Mazzoni, A. The role of matrix metalloproteinases in periodontal disease. Int. J. Environ. Res. Public Health 2020, 17, 4923.
- Franco, C.; Patricia, H.-R.; Timo, S.; Claudia, B.; Marcela, H. Matrix metalloproteinases as regulators of periodontal inflammation. Int. J. Mol. Sci. 2017, 18, 440.
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095.
- Moore, C.S.; Crocker, S.J. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 2012, 180, 12–16.
- Crocker, S.J.; Pagenstecher, A.; Campbell, I.L. The TIMPs tango with MMPs and more in the central nervous system. J. Neurosci. Res. 2004, 75, 1–11.
- Fischer, T.; Riedl, R. Challenges with matrix metalloproteinase inhibition and future drug discovery avenues. Expert Opin. Drug Discov. 2020, 1–14.
- de Bruyn, M.; Vandooren, J.; Ugarte-Berzal, E.; Arijs, I.; Vermeire, S.; Opdenakker, G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 295–358.
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956.
- Alford, V.M.; Kamat, A.; Ren, X.; Kumar, K.; Gan, Q.; Awwa, M.; Tong, M.; Seeliger, M.A.; Cao, J.; Ojima, I.; et al. Targeting the hemopexin-like domain of latent matrix metalloproteinase-9 (proMMP-9) with a small molecule inhibitor prevents the formation of focal adhesion junctions. ACS Chem. Biol. 2017, 12, 2788–2803.
- Houghton, A.M.; Hartzell, W.O.; Robbins, C.S.; Gomis-Rüth, F.X.; Shapiro, S.D. Macrophage elastase kills bacteria within murine macrophages. Nature 2009, 460, 637–641.
- Manka, S.W.; Carafoli, R.; Visse, R.; Bihan, D.; Raynal, N.; Farndale, R.W.; Murphy, G.; Enghild, J.J.; Hohenester, E.; Nagase, H. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. USA 2012, 109, 12461–12466.
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927.
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendörfer, M.; Brandstetter, H. Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079.
- Hariono, M.; Nuwarda, R.F.; Yusuf, M.; Rollando, R.; Jenie, R.I.; Al-Najjar, B.; Julianus, J.; Putra, K.C.; Nugroho, E.S.; Wisnumurti, Y.K.; et al. Arylamide as Potential Selective Inhibitor for Matrix Metalloproteinase 9 (MMP9): Design, Synthesis, Biological Evaluation, and Molecular Modeling. J. Chem. Inf. Model. 2019, 60, 349–359.
- Feinberg, T.Y.; Zheng, H.; Lui, R.; Wicha, M.S.; Yu, S.M.; Weiss, S.J. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 2018, 47, 145–160.
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012, 72, 2339–2349.
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177.
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1242–1285.
- Levin, M.; Udi, Y.; Solomonov, I.; Siga, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1927–1939.
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946.
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974.


