 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Mavromoustakos | + 5222 word(s) | 5222 | 2020-12-31 07:43:57 | | | |
| 2 | Rita Xu | -1519 word(s) | 3703 | 2021-01-06 04:08:08 | | |
Video Upload Options
Antagonists of the AT1receptor (AT1R) are beneficial molecules that can prevent the peptide hormone angiotensin II from binding and activating the specific receptor causing hypertension in pathological states.
1. Introduction
Angiotensin II Type 1 receptor (AT1R) antagonists are a class of molecules that antagonize the detrimental action of the octapeptide hormone Angiotensin II (AII) that acts on the G protein-coupled receptor (GPCR) AT1 and induces hypertension [1][2]. The first drug that was developed, acting as an AT1R antagonist was losartan. After that, the class of molecules targeting AT1R is named SARTANs. Sartans are well known molecules, widely prescribed for the treatment of hypertension and a variety of different diseases [3][4][5][6]. The molecular target of AT1R antagonists has been recently established by the crystallization of these molecules in the receptor’s active site. However, their mechanism of action still remains unknown [7][8]. These molecules could exert their action either directly on the AT1R or indirectly, through a two-step mechanism where membrane bilayers play a significant role in molecule’s binding to the receptor. According to the aforementioned mechanism of action, sartans are initially embedded in the lipid bilayers and then they are diffused in the active site of the receptor [9][10][11][12][13][14].
Our laboratory is actively involved in the comprehensive understanding of the molecular and mechanistic basis of the action of different sartans through the application of a battery of biophysical techniques. Existing evidence is still not unequivocal as far as differentiating between the two mechanisms of action [15][16]. Discovering the exact mechanism that the molecules use to approach their receptors could be an extremely important tool in the drug design process. The design of novel molecules should take into account, not only the binding affinity of the drugs to their receptors, but also their capacity to penetrate lipid bilayers or “survive” in the aquatic environment [17]. Moreover, many obstacles like the accumulation of drug molecules into the cellular membrane [18] could be surpassed by observing the mechanism each drug utilizes to reach its receptor’s active site. In order to unveil the molecular basis of drug’s action and improve its efficiency, exploring the role of membranes in drugs’ binding could provide insightful information for the stereoelectronical requirements of novel drugs. Scientific groups worldwide seek for identifying the potent role of the cellular membrane in the drug binding to a variety of different receptors [9]. According to Paul D. Dobson et al., the biophysical forces that govern the interaction of drugs with lipid membranes are similar to those involved in their interaction with proteins, thus it would be a mistake to ignore them [19]. An important tool to investigate such interactions is NMR spectroscopy. Makriyannis et al. propose a combination of 2H solid state (ss) NMR and small-angle X-ray diffraction in order to obtain the atomic details of the binding between lipophilic cannabinergic ligands and lipid bilayers [10][20]. Drug design targeting the AT1R (GPC receptor [21][22]) is one of the most popular cases where the role of the lipid bilayers has been investigated. Important information should be revealed from such studies so as to lead the drug design process towards more pharmacologically active compounds [23].
2. Conformational Properties, Drug Action and Stability of AT1R Antagonists in Solvents
An important aspect to design novel therapeutic drugs against hypertension is the investigation of the endogenous ligand’s binding to the receptor and the extensive study of its interactions with critical residues on the receptor’s cavity. NMR is a powerful tool to study atomic interactions, hence it could provide very fruitful information concerning the important interactions of the binding. In 2002, D’Amelio et al. conducted 2D NOESY experiments in order to monitor angiotensin’s binding to a specific region of AT1R, the fragment peptide 300–320 (fCT300–320) of the rat angiotensin II receptor AT1a. The NOE effects observed for angiotensin II in complex with AT1R’s fragment were quite different in comparison with the NOEs of angiontensin alone in solution. Moreover, D’ Amelio’s research group indicated that the aromatic residues of angiontensin molecule play a key role in the formation of AT1R-angiontesin II complex. These findings observed by 2D NOESY NMR were further investigated by the chemical shift index (CSI) method and circular dichroism (CD) experiments, revealing the potent conformational helical changes of the receptor, after ligand’s binding [24].
In 1999 our scientific group initiated a research project concerning the conformational properties of losartan and its peptide antagonist, sarmesin, in order to unveil potent common pharmacophore features of the two AT1R antagonists. Indeed, superimposition of the two molecules revealed that losartan indicates some common structural features with sarmesin (i.e., C-terminal amino acids Ile5-His6-Pro7-Phe8-COO- of sarmesin and the corresponding alkyl chain, hydroxyl methyl imidazole, spacer phenyl ring and phenyl tetrazole groups of losartan). This finding explained the rational design of losartan which is based on mimicking the C-terminal of AII [17]. The most important finding in this work was that losartan exists in two possible bioactive enantiomeric conformers as illustrated in Figure 1. However, the biologically important conformation between these two has not yet been identified. This identification is a hard task since these are enantiomeric conformers that cannot be individually evaluated in vitro.
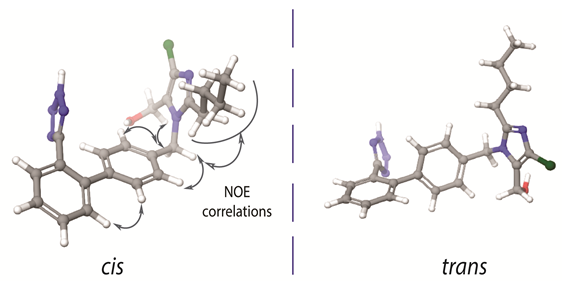
Figure 1. Cis and trans conformers of losartan. The nuclear Overhauser effect (NOE) between the two aromatic rings depicts absence of their orthogonality. (left) The imidazole ring and the tetrazole ring are placed in the same side of the molecule, with respect to the biphenyl ring spacer. (right) The imidazole ring and the tetrazole ring are placed in the opposite side of the molecule, with respect to the biphenyl ring spacer.
There are some important spatial characteristic features in losartan, that have been revealed by 2D NOESY NMR spectroscopy. As it is well known 2D NOESY provides the spatial correlations between protons and it is an essential experiment in order to study the conformational properties of any molecule in a specific environment. The important nuclear Overhauser effect (NOE) spatial correlations are reported for the one of the two possible bioactive conformers illustrated in Figure 1. The spatial vicinity of the n-butyl chain and the spacer phenyl ring indicates that the hydrophobic chain is oriented above the spacer phenyl ring in order to maximize the hydrophobic interactions of the molecule. NOEs could not be differentiated neither when the imidazole and tetrazole rings are placed in the opposite direction (defined as trans) nor when placed in the same direction (defined as cis) with respect to the phenyl ring spacer (Figure 1). Although theoretical calculations indicate the favored formation of the trans conformer, it has not been yet established if this conformation is adopted into a lipid bilayer or in the receptor’s active site. In order to resolve this important issue our laboratory performs Molecular Dynamics (MD) studies so as to establish the structural preference for the two sartans’ conformers both in the lipid bilayers and in the active site of the receptor. According to a recent publication by our research group, candesartan, another sartan molecule, shows distinct preference to adopt a cis conformation in lipid bilayers’ environment [16]. Furthermore, another crucial insight into losartan’s bioactive conformation is an observed NOE between the two spacer phenyl rings which prohibits the orthogonality of the two phenyl groups of the biphenyl tetrazole ring system (Figure 1). Indeed, theoretical calculations have shown that the two phenyl rings are deviating by approximately 60o from the co-planar position. These findings triggered our interest to study the conformational properties of other commercially available AT1R antagonists. Similar conformational features were observed for the AT1R antagonists eprosartan and telmisartan [25][26][27].
Among the AT1R commercial antagonists studied was valsartan. Interestingly, this molecule showed that due to the existence of an amide bond, it could exist in two distinct and almost isoenergetic cis and trans conformations.
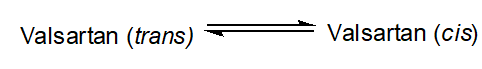
In DMSO solvent we discovered that the trans:cis ratio is 60:40%. Fangand his colleagues used 1H chemical shift analysis, proton relaxation rates and self-diffusion coefficient measurements in order to study the conformation of valsartan when embedded in sodium dodecyl sulfate (SDS) [28]. They observed that the trans conformation is favored versus the cis conformation (Figure 2). These results are in accordance with the results published by our research group, derived from high resolution solution NMR spectroscopy and in silico docking studies. In addition, the combination of NOE measurements and MD simulations showed that valsartan’s biphenyl rings and butyl chain exerted hydrophobic interactions with the alkyl group of SDS. Interestingly, their results revealed that the biological membrane plays an essential role in stabilizing the active state of valsartan. The understanding of the interactions between the sartan drugs and the membrane environment should facilitate the functional mechanism of these compounds with their receptor. The key role of the cellular membrane in sartans’ binding to transmembrane AT1 receptor indicates that the investigation of sartans should also involve AT1R:membrane interactions’ studies. They could also provide insight into the development of new approaches for drug discovery as these were performed by our groups and will be analyzed below.
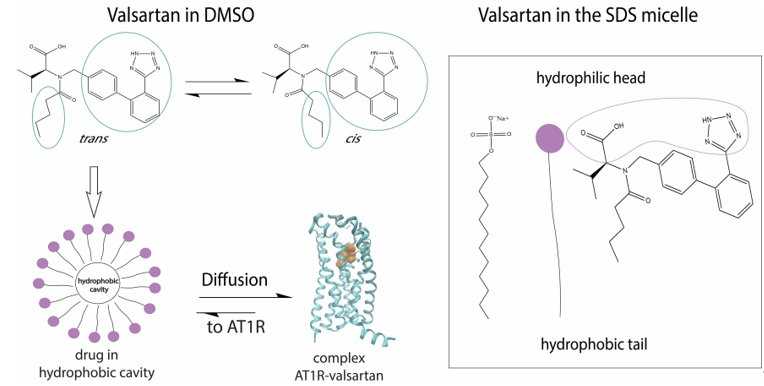
Figure 2. Mechanism of action of valsartan. The trans conformation of the drug is firstly encapsulated in the micelle and then diffused towards AT1R.
Another computational investigation concerning the bioactive conformers of valsartan suggests that the molecule adopts its cis conformation in the receptor’s active site. This particular study aims to observe the changes of AT1R’s conformation when cis or trans valsartan enters the cavity. According to the MD simulations performed, trans conformation stabilizes the structure and locks AT1R in an inactive state, while the cis conformation causes the displacement of an a-helix of AT1R resulting in the expansion of the binding pocket. This extended binding pocket could serve as a potent binding point for the G-protein [29]. Similar helical displacement has been also observed by Khuraijam Dhanachandra Singh et al., when studying angiontensin’s II activation of AT1R. This molecular dynamics study suggests that a van der Waals interaction between Phe8 of angiotensin II and Ile288 of AT1R leads to a sequence of conformational changes in AT1R, resulting in the formation of G-protein binding point and the exposure of a receptor’s helix to cytoplasm [30].
The determination of the absolute conformation of the drugs targeting AT1R through 2D NOESY experiments has been also applied to 6-substituted aminocarbonylbenzimidazole derivatives [31] aiming to inhibit AT1R’s action.
An important fact indicated by the application of NMR is that olmesartan is not stable in CD3OH solvent. The 1H NMR spectrum of olmesartan was altered due to its conversion to its methylated analog with time as confirmed also by mass spectrometry (MS) [32]. This observation led us to develop a simple synthesis for the methylated analog of olmesartan and to evaluate its biological activity. Interestingly, the methylated analogue was almost equipotent with olmesartan depicting that even a relevant biotransformation that could happen in the body, i.e., enzymatically via methyl transferases, will not significantly modify the bioactivity profile of olmesartan (Figure 3).
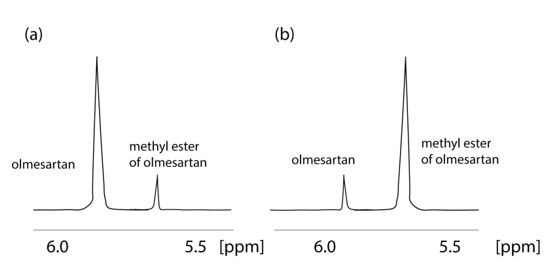
Figure 3. Formation of methyl ether of οlmesartan with time. 1H NMR spectra of 2 mg of olmesartan dissolved in CD3OD obtained on a Bruker Avance III spectrometer (Bruker Biospin GmbH, Reinsteten, Germany) operating at 600.11 MHz for 1H at 45 °C: (a) recorded 4 hours after dissolution; (b) recorded after 52 months.
3. AT1R Antagonists: Membrane Bilayer Interactions
Three angiotensin II peptide antagonists ([Sar1]-AII(1-7)-NH2,[Sar1,Val5,Ala8]-AII and the AII antipeptide, [Glu1,Gly2,Val5,Val8]-AII) have been assessed by NMR and molecular docking studies in a lipid medium in order to determine the conformational requirements of the molecules for their AT1R binding. Under this study, it has been unveiled that histidine residue (6th residue) carries a pivotal role in the determination of the conformation of the peptide antagonists. The insightful NOEs obtained by these studies revealed the interactions between residues 1 and 6, when the antagonists were embedded in micelle solution. 1D and 2D 1H NMR spectra were obtained at different temperatures between 30 to 50 °C. Wilkes et al. discovered that the amide protons associated with residues 4 and 5 are more shielded from the aqueous environment than others participating in the peptide sequence [33].
As it has been already mentioned the exact mechanism of how the AT1R antagonists approach the receptor has not been unveiled and there are two potent scenarios concerning the drug binding mechanism of sartans to the AT1R. According to these theories, either sartans act directly on the AT1R or they exert their action through a two-step mechanism that involves their incorporation into the lipid bilayers and then their diffusion to the active site of the receptor.
13C Cross-polarization and magic-angle spinning (CP/MAS) is a tremendously useful technique used to explore drug:membrane interactions, since the putative incorporation of the molecule into the membrane could be detected by observing the drug’s peaks in the spectra. Chemical shift’s changes and broadening of drug’s peaks can reveal very useful information about the dynamic properties of the molecules into the lipid bilayers. Anisotropic interactions between nuclei, which are usually averaged by Brownian motion in liquid samples, cause significant line broadening in solid state NMR, but they can be averaged to zero by spinning the sample very rapidly at the magic angle (54.74° with respect to the direction of the magnetic field). Cross-polarization (CP) is the most important signal enhancement technique in solid-state NMR. Polarization is first transferred from the abundant spins typically 1H, to the dilute spins typically 13C and the signal from the dilute spins C is then observed. The following 13CP/MAS spectra of the hydrophobic region of dipalmitoylphospatidylcholine (DPPC), DPPC/cholesterol, DPPC/olmesartan and DPPC/cholesterol/olmesartan spectra shows the direct incorporation of cholesterol, since new peaks arise due to its presence. The direct presence of olmesartan in the bilayers is also detected due to the changes it causes to the chemical shifts of DPPC bilayers in this hydrophobic region.It should be notated that olmesartan does not influence the DPPC/cholesterol chemical shifts (Figure 4). This is an intriguing effect as previous studies have shown that the AT1R antagonist losartan does not prefer lipid rafts containing cholesterol. It is evident that AT1R antagonists, in case they act through the mechanism involving lipid bilayers, they do not tend to approach the cholesterol regions of the membrane [34][35].
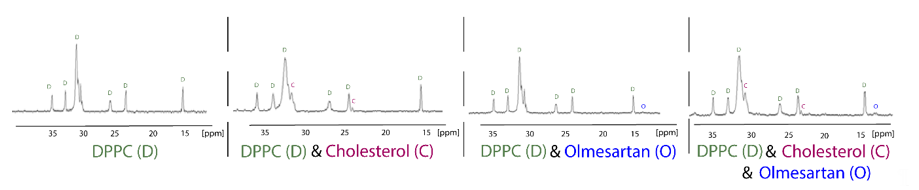
Figure 4. 13C CP/MAS obtained at liquid crystalline phase of DPPC bilayers (45 °C) of the samples DPPC, DPPC/cholesterol (85:15), DPPC/olmesartan (80:20) and (DPPC/cholesterol)/olmesartan (80:20). 13C NMR-spectra were obtained at 150.80 MHz with a 600 MHz Varian spectrometer (Palo Alto, CA) and spinning rate 5 kHz. Sample preparation involved appropriate amounts of DPPC, olmesartan and cholesterol, diluted in chloroform/methanol mixed, dried under stream of argon and then stored under high vacuum overnight. Approximately 50 mg of the sample packed tightly into a zirconia rotor [35]. Reprinted from Biochimica et Biophysica Acta (BBA)—Biomembranes, 1808, Dimitrios Ntountaniotis, Gregor Mali, Simona Golic Grdadolnik, Halabalaki Maria, Alexios-Leandros Skaltsounis, Constantinos Potamitis, Eleni Siapi, Petros Chatzigeorgiou, Michael Rappolt, Thomas Mavromoustakos, Thermal, dynamic and structural properties of drug AT1 antagonist olmesartan in lipid bilayers, pages. 2995–3006, Copyright (2011), with permission from Elsevier.
Hydrated undecoupled 31P NMR spectra of the non-oriented and unsonicated hydrated phospholipid bilayers indicates one broad line that covers approximately a 50 ppm line width. The line shape of the observed powder-type spectrum is attributed to chemical shift anisotropy and proton-phosphorus dipolar interactions [36][37]. The 31P-1H dipolar broadening is removed by 1H decoupling, either in a single free induction decay experiment or in a cross-polarization experiment invented by Hartmann and Hahn in 1962 [38]. 31P is one of the most useful nuclei as it exhibits 6.6% sensitivity relatively to 1H. 31P NMR is a very powerful tool widely applied in model and biological membranes [39].
In 2009 we established a cross-polarization (CP) 31P NMR broadline simulation methodology for studying the effects of drugs in phospholipid bilayers. Based on seven-parameter fittings, this methodology provided information concerning the conformational changes and dynamic effects of drugs in the polar regions of phospholipid bilayers [40]. A simplified dynamical model was used, with the studied dipalmitoylphosphatidyl choline (DPPC) multilamellar bilayers that were considered to be immobilized in the time-scale of the NMR experiment, whereas the lipid molecules were assumed to perform fast overall rotational diffusion in both the liquid crystalline and in the more organized gel phase. A detailed theory of the broadline CP 31P NMR simulations of fully hydrated DPPC dispersions in the form of lipid bilayers has been published earlier [41]. 13CP/MAS experimental indicated that AT1R antagonists indeed affect the DPPC head-group. In order to get a deeper understanding, 31P static NMR spectroscopy was applied. The establishment of this methodology was achieved using losartan. In a subsequent study the prodrug of candesartan, candesartan cilexitil, has been also used as an example. These spectra pinpointed that the prodrug candesartan cilexitil modifies the isotropic shift towards lower values and abolishes the deep minimum observed in the 31P broadline spectra in the gel phase (Figure 5). More detailed information of the techniques and the plethora of information can be found in our previous research work [42].
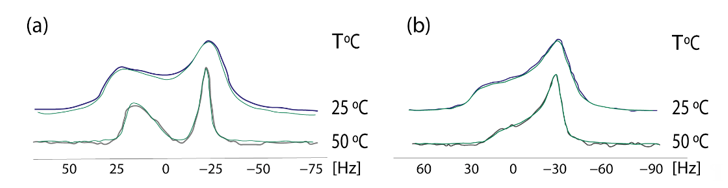
Figure 5. Experimental (black and purple lines) and simulated (green lines) of 31P NMR Scheme 0. (a) DPPC alome (b) DPPC with candesartan cilexetil at the temperatures of 25 and 50 °C. Solid state 31P CP NMR. Spectra were obtained on a Bruker MSL-400 NMR spectrometer (Rheinstetten, Germany) operating at 161.977 MHz using high-power 1H-decoupling. Each spectrum was an accumulation of 1000 scans. The standard CP pulse sequence of the Bruker library was used with the following acquisition parameters: recycling delay 4 s, contact time 5 ms, acquisition time 1 ms, and π/2 pulse for proton 7 ms. The contact time was chosen to give optimal spectra after testing at 1, 3, and 5 ms. The sample was revolved in a 4-mM rotor at a low frequency of 25 Hz. 31P resonance was referenced to H3PO4 (85% in D2O). All simulations were obtained imposing Lorentzian spin packets and automated fitting using the downhill simplex algorithm with a convergence criterion of 0.01 [43]. Reprinted from Biochimica et Biophysica Acta (BBA)—Biomembranes, 1818, Charalambos Fotakis, Grigorios Megariotis, Dionysios Christodouleas, Eftichia Kritsi, Panagiotis Zoumpoulakis, Dimitrios Ntountaniotis, Maria Zervou, Constantinos Potamitis,Aden Hodzic, Georg Pabst,Michael Rappolt,Gregor Mali,Johanna Baldus, Clemens Glaubitz, Manthos G. Papadopoulos, Antreas Afantitis, Georgia Melagraki, Thomas Mavromoustakos, Comparative study of the AT1 receptor prodrug antagonist candesartan cilexetil with other sartans on the interactions with membrane bilayers, pages. 3107–3120 Copyright (2012), with permission from Elsevier.
Solid state 2H NMR is also a valuable tool for getting information on the dynamic properties of a drug embedded in the membrane bilayers. The experiment utilizes the quadrupole echo pulse sequence ((π/2)x − τ − (π/2)y). Thus, the pulse sequence consists of a pair of 90°-phase-shifted π/2 pulses separated by time τ. Due to the interaction between the electric quadrupole moment and the electric field gradient at the nuclear site, the deuterium nucleus gives rise to a doublet. The anisotropic nature of this interaction gives two spectral lines having a frequency difference of ν1 − ν2 = (3/4)ΑQ (3cos2θ − 1), which depends on the angle θ between the magnetic field and the axis of molecular ordering. In the aqueous phospholipid bilayers in the liquid crystalline phase all values of θ are possible and the spectrum consists of two principal peaks that correspond to θ = 90° (perpendicular edges) and two shoulders to θ = 0° (parallel edges). The separation between the two 90°-edges is defined as the quadrupolar splitting (ΔνQ). At temperatures below the main phase transition, the axial diffusion rate is slow or intermediate and appears a conical or rounded spectrum which does not show sharp features. The study of the spectral shape as a function of temperature and the changes in ΔvQ because of the drug incorporation, delivers information on the dynamic properties of the lipid bilayers in the specific labeled region.
The effects of olmesartan in the DPPC bilayers were studied using solid state stationary 2H NMR spectroscopy. It can be observed that in the gel phase, up to 30 °C, the spectrum is featureless. Above this temperature, the dynamics of the lipid bilayers allows the appearance of quadrupolar splittings but still not quite evident to measure. This is indicative that DPPC bilayers are in the ripple phase. In the liquid crystalline phase (45 °C), the spectrum of DPPC bilayers shows three quadrupolar splittings 11,9 (2-S), 17,2 (2-R) and 26,2 (-CD2) kHz [44]. The presence of olmesartan causes a significant increase of ΔνQ at 2′-S and a decrease at 2′-R Thus, this position has a bimodal effect. The spectra in the temperature range of 15–45 °C (Figure 6) pinpoints that between 15 and 30 °C the preparation containing olmesartan is in the gel phase and between 35 and 45 °C it is altered to the liquid crystalline phase. The presence of olmesartan abolishes the pre-transition state.
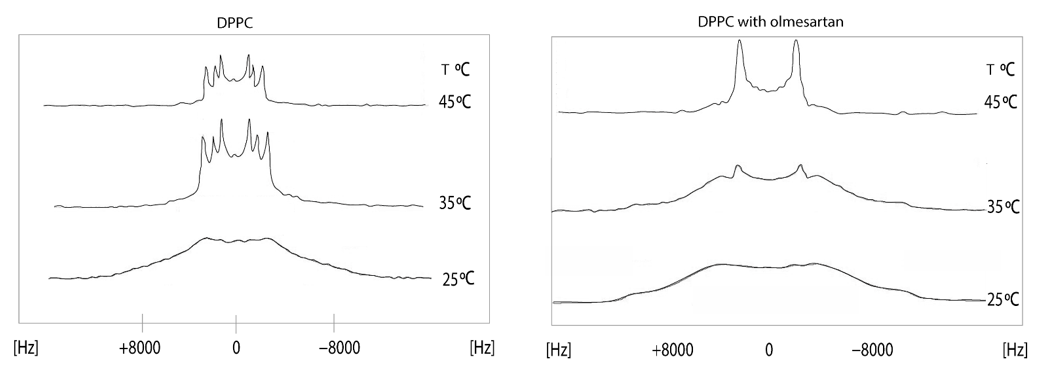
Figure 6. 2Η NMR spectra of 1,2[2,2-2H]-DPPC bilayer (left) at a temperature range of 15–45 °C and 1,2 [2,2-2H]-DPPC bilayer with olmesartan (20% molar ratio) was added (right). 2H solid-state NMR spectra were recorded on a BrukerAvance 600 MHz WB spectrometer equipped with a 4 mm HX double resonance MAS probe. Spectra were recorded without spinning the 4 mm MAS rotor using a solid echo sequence with an echo delay of 35 μs and a pulse length of 2.5 μs [45]. Reprinted from Biochimica et Biophysica Acta (BBA)—Biomembranes, 1838, Dimitrios Ntountaniotis, Tahsin Kellici, Andreas Tzakos, Pinelopi Kolokotroni, Theodore Tselios, Johanna Becker-Baldus, Clemens Glaubitz, Sonyan Lin, Alexandros Makriyannis, Thomas Mavromoustakos, The application of solid-state NMR spectroscopy to study candesartan cilexetil (TCV-116) membrane interactions. Comparative study with the AT1R antagonist drug olmesartan, pages. 2439–2450, Copyright (2014), with permission from Elsevier.
Similar spectra can be also obtained for other labeling schemes at different positions and thus this could reveal the selectivity of the drug to participate in specific topographic position in DPPC bilayers. In addition, comparative studies with other prodrugs or AT1R antagonists can be established using specific deuteration [45].
Another aspect that should be investigated in order to detect the preference of sartans for lipid bilayers should be the ionization states and the pKa of each sartan. А Quantum Mechanics (QM) study on ionization of three sartans (losartan, irbesartan and valsartan) in aqueous media and on interactions with surfactant micelles has been performed by Popović-Nikolić et al. This study suggested that sartans in their amphiprotic form interact predominantly with michelle surfaces, while presenting quite high affinity with the lipid area. Similar results have been acquired for their diprotic acid’s form, but under this protonation state sartans do not interact stongly with the micelle [46].
References
- Ettehad, D.; Emdin, C.A.; Kiran, A.; Anderson, S.G.; Callender, T.; Emberson, J.; Chalmers, J.; Rodgers, A.; Rahimi, K. Blood pressure lowering for prevention of cardiovascular disease and death: A systematic review and meta-analysis. Lancet 2016, 387, 957–967, doi:10.1016/S0140-6736(15)01225-8.
- Wagenaar, L.J.; Voors, A.A.; Buikema, H.; Van Buiten, A.; Lübeck, R.H.; Boonstra, P.W.; Van Veldhuisen, D.J.; Van Gilst, W.H. Functional antagonism of different angiotensin II type I receptor blockers in human arteries. Drugs Ther. 2002, 16, 311–316, doi:10.1023/A:1021729909456.
- Mendis, S.; Puska, P.; Norrving, B. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011; ISBN 978 92 4 156437 3.
- Kellici, T.F.; Tzakos, A.G.; Mavromoustakos, T. Rational drug design and synthesis of molecules targeting the angiotensin II type 1 and type 2 receptors. Molecules 2015, 20, 3868–3897, doi:10.3390/molecules20033868.
- Kellici, T.F.; Liapakis, G.; Tzakos, A.G.; Mavromoustakos, T. Pharmaceutical compositions for antihypertensive treatments: A patent review. Expert Opin. Ther. Pat. 2015, 25, 1–12, doi:10.1517/13543776.2015.1086337.
- Mavromoustakos, T.; Agelis, G.; Durdagi, S. AT1 antagonists: A patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 1–12, doi:10.1517/13543776.2013.830104.
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 2019, 176, 479–490.
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–833, doi:10.1016/j.cell.2015.04.011.
- Vauquelin, G.; Packeu, A. Ligands, their receptors and . plasma membranes. Cell. Endocrinol. 2009, 311, 1–10, doi:10.1016/j.mce.2009.07.022.
- Makriyannis, A.; Tian, X.; Guo, J. How lipophilic cannabinergic ligands reach their receptor sites. Prostaglandins Other Lipid Mediat. 2005, 77, 210–218, doi:10.1016/j.prostaglandins.2004.01.010.
- Mason, R.P.; Rhodes, D.G.; Herbette, L.G.; Mason, R.P.; Mason, R.P.; Rhodes, D.G.; Herbette, L.G.; Herbette, L.G.; Herbette, L.G. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. Med. Chem. 1991, 34, 869–877, doi:10.1021/jm00107a001.
- Mavromoustakos, T.; Yang, D.P.; Makriyannis, A. Small angle X-ray diffraction and differential scanning calorimetric studies on O-methyl-(-)-Δ8-tetrahydrocannabinol and its 5′ iodinated derivative in membrane bilayers. BBA-Biomembr. 1995, 1237, 183–8, doi:10.1016/0005-2736(95)00101-8.
- Mavromoustakos, T.; Zoumpoulakis, P.; Kyrikou, I.; Zoga, A.; Siapi, E.; Zervou, M.; Daliani, I.; Dimitriou, D.; Pitsas, A.; Kamoutsis, C.; et al. Efforts to understand the molecular basis of hypertension through drug: Membrane interactions. Top. Med. Chem. 2005, 4, 445–459, doi:10.2174/1568026043451339.
- Rhodes, D.G.; Sarmiento, J.G.; Herbette, L.G. Kinetics of binding of membrane-active drugs to receptor sites. Diffusion-limited rates for a membrane bilayer approach of 1,4-dihydropyridine calcium channel antagonists to their active site. Pharmacol. 1985, 27, 612–623.
- Zoumpoulakis, P.; Daliani, I.; Zervou, M.; Kyrikou, I.; Siapi, E.; Lamprinidis, G.; Mikros, E.; Mavromoustakos, T. Losartan’s molecular basis of interaction with membranes and AT 1 receptor. Phys. Lipids 2003, 125, 13–25, doi:10.1016/S0009-3084(03)00053-7.
- Kiriakidi, S.; Chatzigiannis, C.; Papaemmanouil, C.; Tzakos, A.G.; Mavromoustakos, T. Exploring the role of the membrane bilayer in the recognition of candesartan by its GPCR AT1 recepto Biochim. Biophys. Acta Biomembr. 2020, 1862, 183142, doi:10.1016/j.bbamem.2019.183142.
- Balaz, S. Does transbilayer diffusion have a role in membrane transport of drugs? Drug Discov. Today 2012, 17, 1079–1087, doi:10.1016/j.drudis.2012.06.003.
- Mayne, C.G.; Arcario, M.J.; Mahinthichaichan, P.; Baylon, J.L.; Vermaas, J.V.; Navidpour, L.; Wen, P.C.; Thangapandian, S.; Tajkhorshid, E. The cellular membrane as a mediator for small molecule interaction with membrane proteins. Biophys. Acta Biomembr. 2016, 1858, 2290–2304, doi:10.1016/j.bbamem.2016.04.016.
- Dobson, P.D.; Kell, D.B. Carrier-mediated cellular uptake of pharmaceutical drugs: An exception or the rule? Rev. Drug Discov. 2008, 7, 205–220, doi:10.1038/nrd2438.
- Tian, X.; Guo, J.; Yao, F.; Yang, D.P.; Makriyannis, A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. Biol. Chem. 2005, 280, 29788–29795, doi:10.1074/jbc.M502925200.
- Szlenk, C.T.; Gc, J.B.; Natesan, S. Does the lipid bilayer orchestrate access and binding of ligands to transmembrane orthosteric/allosteric sites of G protein-coupled receptors? Pharmacol. 2019, 96, 527–541, doi:10.1124/mol.118.115113.
- Odoemelam, C.S.; Percival, B.; Wallis, H.; Chang, M.W.; Ahmad, Z.; Scholey, D.; Burton, E.; Williams, I.H.; Kamerlin, C.L.; Wilson, P.B. G-Protein coupled receptors: Structure and function in drug discovery. RSC Adv. 2020, 10, 36337–36348, doi:10.1039/d0ra08003a.
- Vauquelin, G. Cell membranes… and how long drugs may exert beneficial pharmacological activity in vivo. J. Clin. Pharmacol. 2016, 82, 673–682, doi:10.1111/bcp.12996.
- D’Amelio, N.; Gaggelli, E.; Gaggelli, N.; Lozzi, L.; Neri, P.; Valensin, D.; Valensin, G. Interaction of angiotensin II with the C-terminal 300–320 fragment of the rat angiotensin II receptor AT1a monitored by NMR. Biopolymers 2003, 70, 134–144, doi:10.1002/bip.10426.
- Zoumpoulakis, P.; Grdadolnik, S.G.; Matsoukas, J.; Mavromoustakos, T. Structure elucidation and conformational properties of eprosartan a non peptide Angiotensin II AT1 antagonist. Pharm. Biomed. Anal. 2002, 28, 125–135, doi:10.1016/S0731-7085(01)00603-3.
- Kritsi, E.; Potamitis, C.; Durdagi, S.; Zoumpoulakis, P.; Golic Grdadolnik, S.; Mavromoustakos, T. Molecular insights into the AT1 antagonism based on biophysical and in silico studies of telmisartan. Chem. Res. 2013, 22, 4842–4857, doi:10.1007/s00044-012-0464-5.
- Kellici, T.; Ntountaniotis, D.; Kritsi, E.; Zervou, M.; Zoumpoulakis, P.; Potamitis, C.; Durdagi, S.; Salmas, R.; Ergun, G.; Gokdemir, E.; et al. Leveraging NMR and X-ray data of the free ligands to build better drugs targeting angiotensin II Type 1 G-protein coupled receptor. Med. Chem. 2015, 23, 36–59, doi:10.2174/0929867323666151117122116.
- Li, F.; Wang, L.; Xiao, N.; Yang, M.; Jiang, L.; Liu, M. Dominant conformation of valsartan in sodium dodecyl sulfate micelle environment. Phys. Chem. B 2010, 114, 2719–2727, doi:10.1021/jp908958k.
- Wang, L.; Yan, F. Trans and cis conformations of the antihypertensive drug valsartan respectively lock the inactive and active-like states of angiotensin II type 1 receptor: A molecular dynamics study. Chem. Inf. Model. 2018, 58, 2123–2130, doi:10.1021/acs.jcim.8b00364.
- Singh, K.D.; Unal, H.; Desnoyer, R.; Sadashiva, S.; Foundation, C.C.; States, U. Mechanism of hormone peptide activation of a GPCR: Angiotensin II activated state of AT1R initiated by van der Waals attraction. Chem. Inf. Model. 2019, 59, 373–385, doi:10.1021/acs.jcim.8b00583.Mechanism.
- Wang, J.L.; Zhang, J.; Zhou, Z.M.; Li, Z.H.; Xue, W.Z.; Xu, D.; Hao, L.P.; Han, X.F.; Fei, F.; Liu, T.; et al. Design, synthesis and biological evaluation of 6-substituted aminocarbonyl benzimidazole derivatives as nonpeptidic angiotensin II AT 1 receptor antagonists. J. Med. Chem. 2012, 49, 183–190, doi:10.1016/j.ejmech.2012.01.009.
- Ntountaniotis, D.; Agelis, G.; Resvani, A.; Halabalaki, M.; Liapakis, G.; Spyridaki, K.; Grdadolnik, S.; Merzel, F.; Kostidis, S.; Potamitis, C.; et al. An efficient synthetic method and theoretical calculations of olmesartan methyl ether: Study of biological function of AT1 antagonism. Chem. High Throughput Screen. 2014, 17, 652–662, doi:10.2174/138620731708140922171503.
- Wilkes, B.C.; Masaro, L.; Schiller, P.W.; Carpenter, K.A. Angiotensin II vs. its type I antagonists: Conformational requirements for receptor binding assessed from NMR spectroscopic and receptor docking experiments. Med. Chem. 2002, 45, 4410–4418, doi:10.1021/jm0103155.
- Hodzic, A.; Zoumpoulakis, P.; Pabst, G.; Mavromoustakos, T.; Rappolt, M. Losartan’s affinity to fluid bilayers modulates lipid-cholesterol interactions. Chem. Chem. Phys. 2012, 14, 4780–4788, doi:10.1039/c2cp40134g.
- Ntountaniotis, D.; Mali, G.; Grdadolnik, S.G.; Maria, H.; Skaltsounis, A.L.; Potamitis, C.; Siapi, E.; Chatzigeorgiou, P.; Rappolt, M.; Mavromoustakos, T. Thermal, dynamic and structural properties of drug AT 1 antagonist olmesartan in lipid bilayers. Biophys. Acta Biomembr. 2011, 1808, 2995–3006, doi:10.1016/j.bbamem.2011.08.001.
- Yeagle, P.L. Phosphorus NMR of membranes. In Biological Magnetic Resonance; Springer, Boston, MA, USA, 1990; pp. 1–54.
- McLaughlin, A.C.; Cullis, P.R.; Hemminga, M.A.; Hoult, D.I.; Radda, G.K.; Ritchie, G.A.; Seeley, P.J.; Richards, R.E. Application of 31P NMR to model and biological membrane systems. FEBS Lett. 1975, 57, 213–218.
- Hartmann, S.R.; Hahn, E.L. Nuclear double resonance in the rotating frame. Rev. 1962, 128, 2042, doi:10.1103/PhysRev.128.2042.
- Mavromoustakos, T.; Theodoropoulou, E.; Dimitriou, C.; Matsoukas, J.M.; Panagiotopoulos, D.; Makriyannis, A. Interactions of angiotensin II with membranes using a combination of differential scanning calorimetry and 31P NMR spectroscopy. Pept. Sci. 1996, 3, 175–180, doi:10.1007/BF00128103.
- Fotakis, C.; Christodouleas, D.; Chatzigeorgiou, P.; Zervou, M.; Benetis, N.P.; Viras, K.; Mavromoustakos, T. Development of a CP 31P NMR broadline simulation methodology for studying the interactions of antihypertensive AT1 antagonist losartan with phospholipid bilayers. J. 2009, 96, 2227–2236, doi:10.1016/j.bpj.2008.11.057.
- Benetis, N.P.; Kyrikou, I.; Zervou, M.; Mavromoustakos, T. Static CP 31P NMR multilamellar bilayer broadlines in the absence and presence of the bioactive dipeptide β-Ala-Tyr or Glu. Phys. 2005, 314, 57–72, doi:10.1016/j.chemphys.2005.01.028.
- Fotakis, C.; Christodouleas, D.; Zoumpoulakis, P.; Kritsi, E.; Benetis, N.P.; Mavromoustakos, T.; Reis, H.; Gili, A.; Papadopoulos, M.G.; Zervou, M. Comparative biophysical studies of sartan class drug molecules losartan and candesartan (CV-11974) with membrane bilayers. Phys. Chem. B 2011, 115, 6180–6192, doi:10.1021/jp110371k.
- Fotakis, C.; Megariotis, G.; Christodouleas, D.; Kritsi, E.; Zoumpoulakis, P.; Ntountaniotis, D.; Zervou, M.; Potamitis, C.; Hodzic, A.; Pabst, G.; et al. Comparative study of the AT1 receptor prodrug antagonist candesartan cilexetil with other sartans on the interactions with membrane bilayers. Biophys. Acta Biomembr. 2012, 1818, 3107–3120, doi:10.1016/j.bbamem.2012.08.009.
- Seelig, A.; Seelig, J. Bilayers of dipalmitoyl-3-sn-phosphatidylcholine. Conformational differences between the fatty acyl chains. BBA Biomembr. 1975, 406, 1–5, doi:10.1016/0005-2736(75)90037-1.
- Ntountaniotis, D.; Kellici, T.; Tzakos, A.; Kolokotroni, P.; Tselios, T.; Becker-Baldus, J.; Glaubitz, C.; Lin, S.; Makriyannis, A.; Mavromoustakos, T. The application of solid-state NMR spectroscopy to study candesartan cilexetil (TCV-116) membrane interactions. Comparative study with the AT1R antagonist drug olmesartan. Biophys. Acta Biomembr. 2014, 1838, 2439–2450, doi:10.1016/j.bbamem.2014.06.003.
- Popović-Nikolić, M.R.; Popović, G.V.; Grujić, M.; Nikolić, K.M.; Agbaba, D.D. A Theoretical study on ionization of Sartans in aqueous media and on interactions with surfactant micelles. Mol. Graph. Model. 2018, 82, 67–73, doi:10.1016/j.jmgm.2018.04.008.

