 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pia Clara Pafundi | + 1965 word(s) | 1965 | 2020-12-29 07:31:04 | | | |
| 2 | Camila Xu | Meta information modification | 1965 | 2021-01-06 09:44:18 | | |
Video Upload Options
Endothelial Dysfunction is a condition of altered metabolism and function of endothelium inducing vascular injury and defective repair.
1. Endothelial Dysfunction
Functionally, ED can be defined as a reduced bioavailability of NO, which affects the impaired response to an endothelium-dependent vasodilator such as acetylcholine.
Endothelium-derived NO not only keeps blood flow, though it also acts as a negative modulator of platelet aggregation, pro-inflammatory gene expression, ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 (vascular cell adhesion molecule 1) production, E-selectin expression, ET-1 synthesis, VSMC proliferation, and lipoprotein oxidation [1]. Thus, ED is characterized by a series of features which goes beyond the hemodynamic dysregulation, including excess production of reactive oxygen species (ROS), enhanced expression of adhesion molecules and inflammatory mediators [2], and increased permeability of vascular endothelium. All of these promote both beginning and progression of atherogenesis [3][4].
ED predictive role on the cardiovascular risk has been largely documented in clinical studies by non-invasive, semi-invasive and invasive techniques measuring ED in humans in situ [5][6]. Besides ED functional measures, circulating levels of adhesion molecules and proinflammatory cytokines have also been used as surrogate markers of endothelial activation and cardiovascular risk [7].
2. Endothelial Dysfunction in Diabetes
The literature extensively supports ED as an important risk factor for the development of T2DM cardiovascular complications [8].
Based on a state of insulin resistance (IR), the interaction of three pathological conditions frequently associated with diabetes (hypertension, dyslipidemia and hyperglycemia), plays a pivotal role in the pathogenesis of the atherosclerotic process [9]. In this scenario hyperglycemia, playing a key role in any complication of diabetes [10], is a leading actor.
Acute hyperglycemia, achieved by intra-arterial infusion of dextrose, has been documented to impair endothelium-dependent vasodilation in healthy humans [11]. Likewise, the acute increase in plasma glucose after administration of oral glucose tolerance test (OGTT) determines, within a 1–2 h time period, a reduction of flow-mediated vasodilation in non-diabetic subjects, with a higher response in individuals with impaired glucose tolerance (IGT), and even more in those with diabetes [12]. A similar harmful effect is likely expected from prolonged and repeated post-prandial hyperglycemias, as it may routinely happen in T2DM. These hyperglycemic spikes may exert a dramatic and long-lasting epigenetic “memory” effect on the endothelial function, as reported in ECs cultured in high glucose and then restored to normoglycemia [13], which suggests transient hyperglycemia as a potential HbA1c–independent risk factor for diabetic complications [14]. A recent study in small mesenteric arteries from healthy and diabetic db/db mice has demonstrated that both acute and chronic exposure to high glucose interfere with local and conducted vasodilation in the resistance vasculature mediated by EDH [15].
Strong accumulating evidence suggests oxidative stress, defined as increased formation of ROS, reactive nitrogen species (RNS), and/or decreased antioxidant potentials, as the cornerstone of ED in the development of diabetic complications [16]. This condition triggers the production of pro-inflammatory cytokines and adhesion molecules responsible of intimal lesions formation [17][18]. Indirectly, some downstream processes (e.g., insulin resistance, formation of oxidized-low density lipoprotein (ox-LDL), inhibition of AMP-protein kinase (AMPK), and adiponectin) contribute to inflammation during the progression of atherosclerosis [19]. In turn, inflammation enhances ROS production, with a consequent arise of a variety of vicious cycles which intertwine each other, thus featuring the pathogenic complexity of the diabetes-accelerated atherosclerosis [20][21]. Moreover, endothelial damage increases albuminuria, both an independent and strong marker of CV risk [22][23]. The mechanisms by which hyperglycemia induces endothelial dysfunction are summarized in Figure 1 and are described in detail in the following paragraphs.
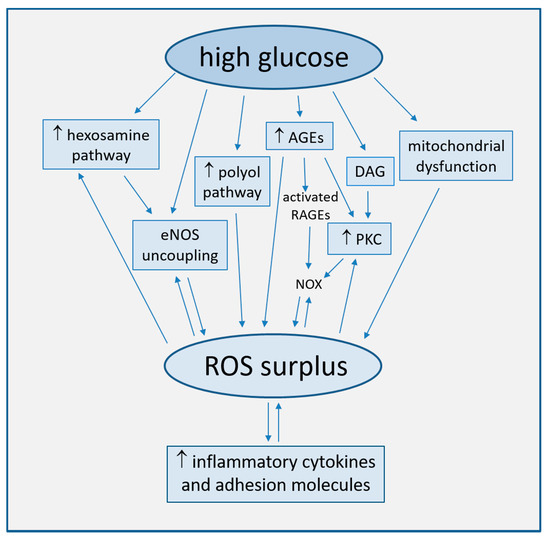
Figure 1. Main mechanisms of high glucose-induced endothelial dysfunction (direct arrows indicate the direction of the pathway, whilst double arrow stands for bidirectional pathway).
2.1. Increased ROS Production
Oxidative stress plays a major role in the pathophysiology of diabetic vascular disease [19][24]. Such a role is consistent with large evidence that increased concentrations of glucose in cultured endothelial cells induce an overproduction of ROS, with the subsequent activation of intracellular signal transduction pathways leading to ED [25][26]. High glucose concentration has been well established to cause endothelial cell damage by both an overproduction of ROS in mitochondria and by multiple biochemical pathways.
2.1.1. Uncoupling of eNOS
The deep reduction in endothelium-dependent vasodilatation associated with T2DM can be linked to changes in eNOS phosphorylation and desensitization induced by signal transduction pathways activated by ROS surplus. As an example, oxidative stress can activate the hexosamine biosynthetic pathway under diabetic and hyperglycemic conditions. This activation is further accompanied by an increase in O-linked N-acetylglucosamine modification of eNOS and a decrease in O-linked serine phosphorylation at residue 1177 [27].
The functional disturbance of the enzyme results in the production of superoxide anion (O2−·) rather than NO, a phenomenon named eNOS uncoupling [28][29].
The ability of eNOS to generate NO can be disabled by the deficiency of tetrahydrobiopterin (BH4), an essential enzyme co-factor, which transforms eNOS into an oxidant-producing enzyme of O2−· [30][31]. ROS may induce oxidative changes of BH4 to dihydrobiopterin (BH2), a BH4 competing compound ineffective as eNOS co-factor. BH2/BH4 competition results in the dissociation of dimeric eNOS to the monomeric form, which acts through its oxygenase domain as an NADPH oxidase, further enhancing ROS generation, in a harmful perpetuation of a vicious circle [1][32][33][34]. Interestingly, the hyperglycemia-induced ED in normal subjects may be prevented by pre-treatment with the BH4 active isomer, 6R-BH4, whilst not by its inactive stereoisomer, 6S-BH4 [35]. In addition, the oral treatment with sepiapterin, a stable precursor of BH4, reduced oxidative stress and improved acetylcholine-mediated endothelium-dependent vasodilation in small mesenteric resistance arteries from db/db obese diabetic mice [36].
GTP cyclohydrolase I (GTPCH I) is the first enzyme in the BH4 biosynthetic pathway, constitutively expressed in endothelial cells and critical for the maintenance of NO synthesis [37]. Studies in HUVECs exposed to high glucose and in streptozotocin-injected diabetic mice have found that hyperglycemia may trigger BH4 deficiency by increasing 26S proteasome-mediated degradation of GTPCH I [38]. This degradation could be either prevented or improved by AMPK overexpression or activation [39].
NO derived from dimeric eNOS and O2−· from monomeric eNOS induces the formation of peroxynitrite (ONOO−·). This may facilitate the release of zinc from the zinc-thiolate cluster of eNOS, which is useful to maintain the dimeric structure of the enzyme, thus resulting in a further enhancement of eNOS uncoupling. Since loss of zinc and eNOS uncoupling activity have been both observed in ECs cells exposed to elevated glucose and in tissues of a diabetic mice model, we may hypothesize a significance of this process under in vivo conditions in diabetes [1][40].
The functions of many proteins may be affected by increased oxidant levels. As an example, a characteristic reaction of ONOO−· is the nitration of protein-bound tyrosine residues to generate 3-nitrotyrosine–positive proteins [41]. Some researchers have suggested that an increased nitration of PGI2 synthase (PGIS), more likely via dysfunctional eNOS, may characterize the diabetic disease. Such a hypothesis stands on observations that exposure of isolated bovine coronary arteries to high glucose switched angiotensin II–stimulated PGI2-dependent relaxation into a persistent vasoconstriction [42]. As well, a significant suppression of PGIS activity, along with increased O2−· and PGIS-nitration, was also observed in aortas of streptozotocin-treated diabetic mice [42].
2.1.2. Mitochondrial Dysfunction
The mitochondrial electron transport chain (ETC) is the primary source of hyperglycemia-induced ROS production via a greater oxygen use, increased redox potential and shift of O2 transport towards the respiratory chain complex II [16][20]. Other mechanisms of mitochondrial dysfunction include increased NADH/FADH2 ratio [43] and mitochondrial fission, which triggers an accumulation of fragmented mitochondria with impaired ETC activity [44].
2.1.3. Activation of the Polyol Pathway
Increased intracellular glucose levels overload ETC and are shunted into alternative pathways, in turn generating ROS. In the polyol pathway, accounting for >30% of glucose metabolism during hyperglycemia [45], glucose is converted by NADPH-dependent aldose-reductase to the sugar alcohol sorbitol, and sorbitol to fructose by sorbitol-dehydrogenase. The oxidative stress generated by these reactions depends on the consumption of NADPH, a cofactor required to regenerate the ROS scavenger glutathione (GSH), and on NAD+ reduction to NADH, which is subsequently oxidized by NADH oxidase, with consequent production of superoxide ions [46]. Aldose-reductase has been indeed implied in the increased expression of inflammatory cytokines [47][48].
2.1.4. Generation of Advanced Glycation End-Products (AGEs)
In conditions of hyperglycemia, the nonenzymatic fragmentation of the glycolytic intermediate triose phosphate produces methylglyoxal, precursor of the majority of AGE products formed by a nonenzymatic reaction of either ketones or aldehydes and the amino groups of proteins, during which large amounts of ROS are generated [16].
AGEs can interact with two types of cell surface receptors, scavengers involved in AGE removal and receptors for AGE (RAGEs), which initiate detrimental cellular signals, promoting inflammation and atherogenesis [20][49][50]. As an example, AGEs dose-dependently activate oxidative stress-mediated P38 activation of mitogen-activated protein kinase (MAPK) signaling in endothelial cells, which enhances NO synthesis inhibition by AGEs [51].
Both AGEs and methylglyoxal also promote the expression of RAGEs ligands. In particular, oxidized AGEs activate RAGEs to stimulate NADPH oxidase (NOX) [52], another important source of ROS production. NOX, which in healthy state determines ROS production, in pathological conditions may be hyper-expressed and hyperactive, as observed in cultured mice microvascular endothelial cells (MMECs) and human umbilical artery endothelial cells (HUAECs) exposed to high glucose [53][54]. Cells exposed to glucose fluctuations produce higher levels of NOX-derived ROS as compared to cells steadily exposed to high glucose, thus indicating the detrimental effect on vascular health of acute glycemic variations [55].
2.1.5. Activation of Protein Kinase C (PKC)
PKC is a serine/threonine related protein kinase acting in a wide variety of biological systems and regulating cell growth and proliferation, senescence, and apoptosis. The enzyme, once activated, induces many atherogenic processes, like ROS overproduction, endothelial dysfunction, increased vascular permeability, and inhibited angiogenesis [24][56].
In particular, NOX PKC-dependent activation is considered among the major sources of high glucose-induced ROS production, even more than mitochondrion [57][58].
In either a hyperglycemic or diabetic environment, PKC is activated by oxidative stress and AGEs and by diacylglycerol (DAG), whose levels increase in endothelial cells due to the shunting of glycolytic intermediates to dihydroxyacetone phosphate [56][59]. DAG-PKC is among the several cellular pathways activating when oxidative stress causes DNA fragmentation and stimulation of the DNA repair enzyme, nuclear poly ADP ribose polymerase (PARP). This enzyme inhibits the glyceraldehyde-3-phosphate dehydrogenase (GAPDH), shunting early glycolytic intermediates into pathogenic signaling pathways, including AGE, polyol, DAG-PKC, and hexosamine pathways [16].
2.2. Endothelial Apoptosis and Senescence
Endothelial cell apoptosis and senescence are pivotal processes for the development of atherosclerosis, due to their activation by a plethora of pathways sharing the common pathophysiological mechanism of oxidative stress [60][61][62].
Studies on cultured ECs have shown that the promotion of senescence features (e.g., shortening of telomere length, elevated DNA damage, increase genomic instability and growth arrest) can be modulated by two factors intrinsically related to diabetes, high glucose [63], and AGE products [64], thus enhancing the intracellular levels of oxidative stress [65][66][67]. The implied cellular signals are diverse. As observed in high glucose exposed umbilical vein endothelial cells (HUVECs), Bax protein expression increases in the absence of Bcl-2 modifications, producing an elevated Bax/Bcl-2 ratio which activates the cleavage of procaspase-3 into active caspase-3, a crucial mediator of apoptosis [68]. As well, also the high-glucose induced NF-kB-dependent activation of c-Jun N-terminal kinase (JNK) and ROS-dependent Akt dephosphorylation may be involved [69].
Intriguingly, carbonic anhydrase, overexpressed in endothelial cells of diabetic ischemic heart, determines endothelial cell apoptosis in vitro, thus playing a key role in the remodeling process [70].
2.3. Other Pathogenetic Mechanisms of Vascular Dysfunction
A dysregulation of microRNAs (miRNAs), small non-coding RNAs, may contribute to the progression of atherosclerosis and diabetes-induced vascular dysfunction. As an example, a reduction in miRNA-126 levels has been associated with an increased leucocyte adherence to ECs and impairment of peripheral angiogenesis in T2DM [71]. Moreover, miR-29c and miR-204 were significantly dysregulated in atherosclerotic plaques from patients with DM [72].
T2DM has been proven as characterized by an imbalance of gut microbiota, which can directly promote atherogenesis by oxidative stress, inflammation, and changes in some metabolites, even though the bacteria possibly associated with progression of diabetes-accelerated atherosclerosis have not been identified yet [20].
References
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Saccomanno, F.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-α. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Ferraraccio, F.; Rizzo, M.R.; Portoghese, M.; Barbieri, M.; Basilio, C.; Nersita, R.; Siniscalchi, L.I.; Sasso, F.C.; Ambrosino, I.; et al. Innate Immune Activity in Plaque of Patients with Untreated andl-Thyroxine-Treated Subclinical Hypothyroidism. J. Clin. Endocrinol. Metab. 2011, 96, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial Dysfunction, Oxidative Stress, and Risk of Cardiovascular Events in Patients with Coronary Artery Disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Phillips, S.A. Assessment and Prognosis of Peripheral Artery Measures of Vascular Function. Prog. Cardiovasc. Dis. 2015, 57, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; DeFelice, A.F.; Hanig, J.P.; Colatsky, T. Biomarkers of Endothelial Cell Activation Serve as Potential Surrogate Markers for Drug-induced Vascular Injury. Toxicol. Pathol. 2010, 38, 856–871. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef]
- Kim, J.-A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Williams, S.B.; Goldfine, A.B.; Timimi, F.K.; Ting, H.H.; Roddy, M.-A.; Simonson, D.C.; Creager, M.A. Acute Hyperglycemia Attenuates Endothelium-Dependent Vasodilation in Humans In Vivo. Circulation 1998, 97, 1695–1701. [Google Scholar] [CrossRef]
- Kawano, H.; Motoyama, T.; Hirashima, O.; Hirai, N.; Miyao, Y.; Sakamoto, T.; Kugiyama, K.; Ogawa, H.; Yasue, H. Hyperglycemia rapidly suppresses flow-mediated endothelium- dependent vasodilation of brachial artery. J. Am. Coll. Cardiol. 1999, 34, 146–154. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Sasso, F.C.; Siniscalchi, M.; Paolisso, P.; Rizzo, M.R.; Ferraro, F.; Stabile, E.; Sorropago, G.; Calabrò, P.; Carbonara, O.; et al. Peri-Procedural Tight Glycemic Control during Early Percutaneous Coronary Intervention Is Associated with a Lower Rate of In-Stent Restenosis in Patients with Acute ST-Elevation Myocardial Infarction. J. Clin. Endocrinol. Metab. 2012, 97, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Lemmey, H.A.L.; Ye, X.; Ding, H.; Triggle, C.R.; Garland, C.J.; Dora, K.A. Hyperglycaemia disrupts conducted vasodilation in the resistance vasculature of db/db mice. Vasc. Pharmacol. 2018, 29–35. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nat. Cell Biol. 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Vascular Adhesion Molecules in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 35. [Google Scholar] [CrossRef]
- Minutolo, R.; Sasso, F.C.; Chiodini, P.; Cianciaruso, B.; Carbonara, O.; Zamboli, P.; Tirino, G.; Pota, A.; Torella, R.; Conte, G.; et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: A comparative analysis in nephrology, diabetology and primary care settings. J. Hypertens. 2006, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Russo, P.D.; Amstad, P.; Cerutti, P. High Glucose Induces Antioxidant Enzymes in Human Endothelial Cells in Culture: Evidence Linking Hyperglycemia and Oxidative Stress. Diabetes 1996, 45, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Förstermann, U.; Munzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Aljofan, M.; Ding, H. High glucose increases expression of cyclooxygenase-2, increases oxidative stress and decreases the generation of nitric oxide in mouse microvessel endothelial cells. J. Cell. Physiol. 2009, 222, 669–675. [Google Scholar] [CrossRef]
- Pannirselvam, M.; Verma, S.; Anderson, T.J.; Triggle, C.R. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db−/−) mice: Role of decreased tetrahydrobiopterin bioavailability. Br. J. Pharmacol. 2002, 136, 255–263. [Google Scholar] [CrossRef]
- Moens, A.L.; Kass, D.A. Tetrahydrobiopterin and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2006, 26, 2439–2444. [Google Scholar] [CrossRef]
- Wever, R.M.; Van Dam, T.; Van Rijn, H.J.; De Groot, F.; Rabelink, T.J. Tetrahydrobiopterin Regulates Superoxide and Nitric Oxide Generation by Recombinant Endothelial Nitric Oxide Synthase. Biochem. Biophys. Res. Commun. 1997, 237, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Tatham, A.L.; Hale, A.B.; Alp, N.J.; Channon, K. Critical Role for Tetrahydrobiopterin Recycling by Dihydrofolate Reductase in Regulation of Endothelial Nitric-oxide Synthase Coupling: Relative importance of the de novo biopterin synthesis versus salvage pathways. J. Biol. Chem. 2009, 284, 28128–28136. [Google Scholar] [CrossRef] [PubMed]
- Channon, K.M. Tetrahydrobiopterin and Nitric Oxide Synthase Recouplers. Bone Regul. Osteoporos. Ther. 2020, 1–14. [Google Scholar] [CrossRef]
- Ihlemann, N.; Rask-Madsen, C.; Perner, A.; Dominguez, H.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am. J. Physiol. Circ. Physiol. 2003, 285, H875–H882. [Google Scholar] [CrossRef]
- Pannirselvam, M.; Simon, V.; Verma, S.; Anderson, T.; Triggle, C.R. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br. J. Pharmacol. 2003, 140, 701–706. [Google Scholar] [CrossRef]
- Franscini, N.; Bächli, E.; Blau, N.; Fischler, M.; Walter, R.B.; Schaffner, A.; Schoedon, G. Functional Tetrahydrobiopterin Synthesis in Human Platelets. Circulation 2004, 110, 186–192. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.-H. Proteasome-Dependent Degradation of Guanosine 5′-Triphosphate Cyclohydrolase I Causes Tetrahydrobiopterin Deficiency in Diabetes Mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.-H. In Vivo Activation of AMP-Activated Protein Kinase Attenuates Diabetes-Enhanced Degradation of GTP Cyclohydrolase I. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hatley, M.E.; Bolick, D.T.; Palmer, L.A.; Edelstein, D.; Brownlee, M.; Hedrick, C.C. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 2004, 47, 1727–1734. [Google Scholar] [CrossRef]
- Wu, N.; Shen, H.; Liu, H.-N.; Wang, Y.; Bai, Y.; Wu, N. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hart, P.C.; Germain, D.; Bonini, M.G. SOD2 and the Mitochondrial UPR: Partners Regulating Cellular Phenotypic Transitions. Trends Biochem. Sci. 2016, 41, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2011, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: A potential target for the prevention of diabetic complications. Pharmacol. Rev. 1998, 50, 21–34. [Google Scholar]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose Reductase, Oxidative Stress, and Diabetic Mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef]
- Reddy, A.B.M.; Ramana, K.V.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Aldose Reductase Regulates High Glucose-Induced Ectodomain Shedding of Tumor Necrosis Factor (TNF)-α via Protein Kinase C-δ and TNF-α Converting Enzyme in Vascular Smooth Muscle Cells. Endocrinology 2008, 150, 63–74. [Google Scholar] [CrossRef]
- Yadav, U.C.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibition suppresses airway inflammation. Chem. Interact. 2011, 191, 339–345. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Shen, C.; Li, Q.; Zhang, Y.C.; Ma, G.; Feng, Y.; Zhu, Q.; Dai, Q.; Chen, Z.; Yao, Y.; Chen, L.; et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed. Pharmacother. 2010, 64, 35–43. [Google Scholar] [CrossRef]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar] [PubMed]
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell. Physiol. 2007, 212, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Taye, A.; Saad, A.H.; Kumar, A.H.; Morawietz, H. Effect of apocynin on NADPH oxidase-mediated oxidative stress-LOX-1-eNOS pathway in human endothelial cells exposed to high glucose. Eur. J. Pharmacol. 2010, 627, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells: The Role of Protein Kinase C and NAD(P)H-Oxidase Activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein Kinase C-Dependent Increase in Reactive Oxygen Species (ROS) Production in Vascular Tissues of Diabetes: Role of Vascular NAD(P)H Oxidase. J. Am. Soc. Nephrol. 2003, 14, 227S–232S. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Xia, P.; Inoguchi, T.; Kern, T.S.; Engerman, R.L.; Oates, P.J.; King, G.L. Characterization of the Mechanism for the Chronic Activation of Diacylglycerol-Protein Kinase C Pathway in Diabetes and Hypergalactosemia. Diabetes 1994, 43, 1122–1129. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E-/- mice. Sci. Rep. 2015, 4, 5519. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Fukuo, K.; Yasuda, O.; Hotta, M.; Miyazaki, J.; Takemura, Y.; Kawamoto, H.; Ichijo, H.; Ogihara, T. Apoptosis Signal-Regulating Kinase 1 Mediates Cellular Senescence Induced by High Glucose in Endothelial Cells. Diabetes 2006, 55, 1660–1665. [Google Scholar] [CrossRef]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated Collagen I Induces Premature Senescence-Like Phenotypic Changes in Endothelial Cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.-S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zou, G.; Gu, J.; Zhang, J. L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res. Clin. Pr. 2010, 89, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Matsui-Hirai, H.; Hayashi, T.; Yamamoto, S.; Ina, K.; Maeda, M.; Kotani, H.; Iguchi, A.; Ignarro, L.J.; Hattori, Y. Dose-Dependent Modulatory Effects of Insulin on Glucose-Induced Endothelial Senescence In Vitro and In Vivo: A Relationship between Telomeres and Nitric Oxide. J. Pharmacol. Exp. Ther. 2011, 337, 591–599. [Google Scholar] [CrossRef]
- Yang, Z.-H.; Mo, X.; Gong, Q.; Pan, Q.; Yang, X.; Cai, W.; Li, C.; Ma, J.-X.; He, Y.; Gao, G. Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis 2008, 13, 1331–1343. [Google Scholar] [CrossRef]
- Ho, F.M.; Lin, W.-W.; Chen, B.C.; Chao, C.M.; Yang, C.-R.; Lin, L.Y.; Lai, C.C.; Liu, S.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell. Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic Anhydrase Activation Is Associated with Worsened Pathological Remodeling in Human Ischemic Diabetic Cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef]
- Tang, N.; Jiang, S.; Yang, Y.; Liu, S.; Ponnusamy, M.; Xin, H.; Yu, T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018, 36, e12436. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Tarallo, R.; Marino, F.; Giurato, G.; Veneziano, C.; Aquila, I.; Scalise, M.; Mancuso, T.; Cianflone, E.; et al. miRNA Regulation of the Hyperproliferative Phenotype of Vascular Smooth Muscle Cells in Diabetes. Diabetes 2018, 67, 2554–2568. [Google Scholar] [CrossRef] [PubMed]

