 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Idalia Garza-Veloz | + 2058 word(s) | 2058 | 2020-12-25 08:33:53 | | | |
| 2 | Vicky Zhou | -2 word(s) | 2056 | 2021-01-05 05:08:52 | | | | |
| 3 | Vicky Zhou | Meta information modification | 2056 | 2021-01-06 04:31:18 | | | | |
| 4 | Margarita L. Martinez-Fierro | Meta information modification | 2056 | 2021-01-06 18:56:51 | | |
Video Upload Options
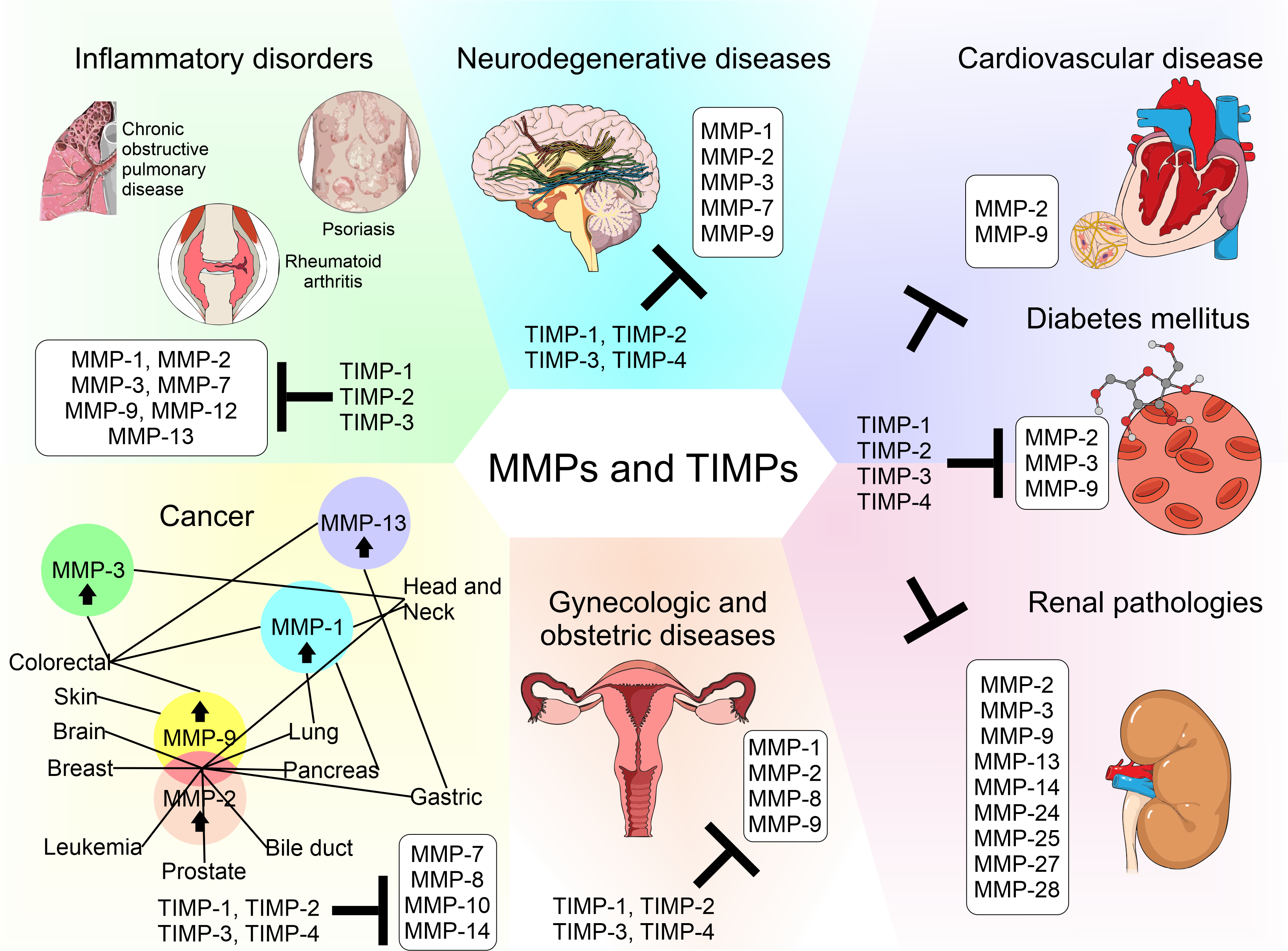
Matrix metalloproteinases (MMPs) are a family of zinc-dependent extracellular matrix (ECM) remodeling endopeptidases that have the capacity to degrade almost every component of the ECM. The degradation of the ECM is of great importance, since it is related to embryonic development and angiogenesis. It is also involved in cell repair and the remodeling of tissues. When the expression of MMPs is altered, it can generate the abnormal degradation of the ECM. This is the initial cause of the development of chronic degenerative diseases and vascular complications generated by diabetes. In addition, this process has an association with neurodegeneration and cancer progression. Within the ECM, the tissue inhibitors of MMPs (TIMPs) inhibit the proteolytic activity of MMPs. TIMPs are important regulators of ECM turnover, tissue remodeling, and cellular behavior. Therefore, TIMPs (similar to MMPs) modulate angiogenesis, cell proliferation, and apoptosis. An interruption in the balance between MMPs and TIMPs has been implicated in the pathophysiology and progression of several diseases.
1. Introduction
The extracellular matrix (ECM) not only plays a supporting role for organs and tissues but also actively participates in other functions, such as regulation of the cell cycle and cell motility, survival, and apoptosis, as well as the distribution of growth factors and integration of signals into cells. The ECM is made up of hundreds of molecules, including proteoglycans; glycosaminoglycans; structural proteins, such as collagen and elastin; adhesion proteins, such as fibronectin and laminin; and proteases called matrix metalloproteases (MMPs) [1]. The MMPs belong to a family of endopeptidases that contains 23 members. These contain zinc, are dependent on calcium, and can degrade and remodel the proteins that form the ECM. They also participate in different biological and physiological processes that are regulated by hormones, growth factors, and cytokines [2]. Based on their sub-cellular distribution and specificity for components of the ECM, the MMPs are divided into membrane-type matrix metalloproteases (MT-MMPs), collagenases, gelatinases, stromelysins, and matrilysins. Collagenases (MMP-1, MMP-8, MMP-13, and MMP-18) degrade triple-helical fibrillar collagen, which is fundamental in bone and ligaments. Gelatinases (MMP-2 and MMP-9) are involved in different cellular process including angiogenesis and neurogenesis; these proteases alter the molecules of the basal lamina, subsequently leading to cell death. Stromelysins (MMP-3, MMP-10, and MMP-11) are small proteases that degrade segments of the ECM. Matrilysins (MMP-7 and MMP-26) process cell surface molecules and digest ECM components. MT-MMPs have collagenolytic activity and may activate some proteases and components of the cell surface [1][3]. MMPs are also classified into eight groups according to their structure. Among these, five are secreted and three are bound to membranes (MT-MMPs) [1]. Some human MMPs display a signal peptide that directs them to the endoplasmic reticulum, the pro-domain (a pro-peptide with a thiol group that interacts with zinc and keeps them as inactive zymogens), and a catalytic domain with a zinc-binding site [1][2]. MMP-23 undergoes type II secretion and therefore does not have an N-terminal signal sequence. Despite most MMPs having N-terminal signal sequences, these do not result in 100% of the protease being secreted; signal sequences can be inefficiently recognized by the sec61 translocon, resulting in a significant fraction of the proteins remaining in the cytoplasm [4][5]. The conservation of an inefficient signal sequence in MMP-2 orthologs suggests the presence of selective pressure to retain a significant fraction of this protease within the cytoplasm. This could support the notion that MMP-2 has important, but unknown, physiological functions within cells.
MMPs are inhibited by tissue inhibitors of MMPs (TIMPs), which are endogenous protein regulators. The TIMP family (TIMP-1–4), are proteins made up of 184–194 amino acids that are ≈21 kDa in molecular weight. The TIMP family has similar but not identical protease inhibitory profiles [6]. TIMPs are present in the ECM in a soluble form, except for TIMP-3, which is bound to the ECM. All TIMPs inhibit MMPs through reversible blockage, forming 1:1 stoichiometric complexes [6]. TIMPs selectively inhibit different MMPs and members of the families Disintegrin and Metalloproteinase (ADAM) and Disintegrin and Metalloproteinase with Thrombospondin motifs (ADAMTS) [6][7]. TIMPs also are important for the activation and uptake/removal of MMPs from the extracellular environment. TIMP function determines the influence of the ECM on cell phenotype, cell adhesion molecules, cytokines, chemokines, and growth factors. They are formed by an amino-terminal domain, which is the inhibiting domain that binds to the active site of MMPs and domain C. The ability of TIMPs to inhibit MMPs is due to their interaction in the terminal N-domain. Domain C gives TIMPs the ability to interact with the hemopexin domain of some MMPs [8].
2. Targeting MMP and TIMP Function (Inhibitors)
The biology of MMPs and the understanding of their regulation led to the development of basic and preclinical research in animal models and clinical research in patients with pathologies including neurological, cardiovascular, gynecological, and inflammatory conditions—mainly cancer. These studies have focused on MMPs, because it is believed that they are the cause of alterations to important physiological mechanisms that lead to abnormal angiogenesis, apoptosis, and immune modulation, therefore contributing to tumor growth and/or metastasis [9]. Research studies funded by the pharmacological industry have begun with the development of broad-spectrum MMP inhibitors, but the precise physiology and biology of MMPs and the details of their evolution through time and regulation by other molecules are unknown. Clinical trials have not had the expected results, worsening the prognosis of diseases, as they were administered in advanced stages, causing adverse effects, such as musculoskeletal toxicity [9]. Nowadays, MMPs are known to have roles in physiological processes such as embryogenesis, angiogenesis, tissue remodeling, bone development, wound healing, mammary involution, cell migration, facilitation of the release of bound signaling molecules, activation of signaling molecules, and immunity [9]. MMP inhibitors and therapies that act against MMPs are listed below.
(a) Hydroxamate-based inhibitors. These molecules were the first MMP inhibitors to be developed. They are based on the structure of collagen and predominantly involve compounds containing a backbone designed to mimic the natural peptide substrate of the desired MMPs and a group that chelates the catalytic Zn2+ ion. They reduce the contribution of the rest of the compound to the inhibitor−enzyme binding process, thereby favoring broad-spectrum inhibition. Some examples of this type of drug are marimastat, ilomastat, and batimastat. Batimastat was the first MMP inhibitor to enter clinical trials, and it was shown to inhibit several MMPs, including MMP-1, MMP-2, MMP-7, and MMP-9, demonstrating antitumor effects in animal models of human ovarian cancer, colorectal cancer, melanoma, and hemangioma. Marimastat was found to be ineffective, and it caused musculoskeletal toxicity in a randomized Phase III trial for metastatic breast cancer cases that were stable or responding after first-line chemotherapy. The cause of this musculoskeletal pain is now thought to be the inhibition of ADAM and ADAMTS family members, including ADAM17. MMP inhibitors have often been administered too late to make a difference, as basic research was done in the early stages of the disease, and clinical research was intended to be a last aid in the treatment of terminal patients [9].
(b) New generation of hydroxamate-based MMP inhibitors. This new generation of MMP inhibitors has been made more specific, in an effort to decrease the adverse effects. Examples of them are MMI-270, MMI-166, PD-166793, ABT-770, cipemastat, and prinomastat. MMI-166 is a selective inhibitor of MMP-2, MMP-9, and MMP-14. PD-166793, prinomastat, and ABT-770 were developed to avoid binding to the “shallow pocket” of MMP-1 based on the idea (at that time) that MMP-1-sparing inhibitors would not induce musculoskeletal toxicity. Cipemastat, which inhibits MMP-1, MMP-3, and MMP-9, was used for the treatment of rheumatoid arthritis and osteoarthritis, but it was not shown to prevent the progression of joint damage in patients. New hydroxamate inhibitors are being developed using structure−activity relationship (SAR) analysis, which can aid in the identification of molecular substructures related to the presence or absence of biological activity [9].
(c) Non-hydroxamate MMP inhibitors. Hydroxamic acids are often metabolically labile, but there are several other zinc-binding groups that are stable. Reverse hydroxamates and non-hydroxamate inhibitors, for example, carboxylates, hydrocarboxylates, sulfhydryls, phosphoric acid derivatives, and hydantoins, were developed to avoid the limitations associated with first-generation MMP inhibitors, such as metabolic inactivation and the chelation of metals of other metalloproteins [9]. The MMP structure has been revealed with the use of crystallography, and new inhibitors have been designed with various peptidomimetic and non-peptidomimetic backbone structures. Rebimastat, a thiol zinc-binding group, is a broad-spectrum MMP inhibitor; however, a Phase II trial in early-stage breast cancer and a Phase III trial in non-small cell lung carcinoma both revealed adverse effects. [9]. Tanomastat was demonstrated to have difficulties with the dosing and timing of administration in relation to disease progression, and the efficacy of this drug was shown to be variable, with contradictory outcomes being obtained depending on the timing of administration [9]. Ro 28-2653 inhibits MT1-MMP, MT3-MMP, MMP-2, MMP-8, and MMP-9 but spares MMP-1 and ADAM17 activity. Several promising animal studies have demonstrated its antitumor and antiangiogenic activity, but they did not progress to clinical trials [9]. Potent inhibitors of MMP-13 and MMP-12 were developed using the alternative zinc-binding group hydantoin. Several biphenyl sulfonamide carboxylate MMP inhibitors were designed to treat osteoarthritis by inhibiting MMP-13. Tetracycline antibiotics, such as doxycycline and minocycline, have innate MMP inhibitory capacity. Doxycycline is indicated for use in periodontal disease and is the only collagenase inhibitor approved by the US Food and Drug Administration for any human disease [9].
(d) Targeting alternative binding sites. To reduce the off-target effects observed in clinical trials and to avoid broad MMP inhibition owing to the high structural homology of the different MMPs, recent research has switched from targeting the catalytic site to targeting alternative, less conserved sites. In addition to the Zn2+ ion in their catalytic sites, MMPs possess subsites (S) designated as unprimed or primed. The P1′−S1′ interaction is the main determinant of the affinity of inhibitors and cleavage positions of peptide substrates. For example, extension of the P1 substituent was used to gain MMP-13 selectivity over the highly homologous MMP-2. The use of NMR and X-ray crystallography methods combined with computational methods enables the modeling of drug−protein interactions with non-hydroxamate inhibitors of MMPs that bind to sites other than the catalytic sites. Several of these inhibitors demonstrated impressive MMP-13 selectivity and resulted in reduced clinical symptoms when tested in mouse models of rheumatoid osteoarthritis and arthritis. The combination of methods with computational prediction revealed hidden sites in the MMP structure, which can be exploited for the rational design of novel molecular effectors and therapeutic agents [9].
(e) Antibody-based therapeutics. These molecules have high selectivity and they possess several functional blocking antibodies. They selectively target the membrane-anchored MMPs. The highly selective antibody-based MMP-14 inhibitor DX-2400 has shown antitumor, antiangiogenic, and anti-invasive properties, and it blocks MMP-14-dependent pro-MMP-2 processing. MMP-14 inhibitory antibodies have been successfully tested in vitro and in vivo. Based on the three-dimensional structure and amino acid sequence of MMP-13, a neutralizing antibody that binds to the active form of MMP-13 but not to the latent form or to other MMPs was developed [9].
(f) Endogenous inhibitors of MMP function. α2-macroglobulin is a large serum protein that regulates MMP activity. MMPs are entrapped within the macroglobulin, thus preventing the MMPs from accessing large substrates. Polymyxin B-conjugated α2-macroglobulin demonstrated protective effects in mouse models of sepsis, and this effect was linked to its binding to, and neutralization of, inflammatory cytokines; MMP inhibition has not been studied [9].
TIMPs could theoretically form the basis of another novel class of MMP inhibitors, and they have rarely been considered. They have been used in model systems to yield vital clues about the efficacy of MMP inhibitors in diseases. However, TIMPs not only inhibit MMPs but, in specific cases, they can even indirectly promote MMP activity. For example, domain-specific overexpression of TIMP-2 and TIMP-3 revealed the MMP-independent functions of TIMPs during development [9].
Although TIMPs have been considered in current therapeutic strategies for the regulation of MMPs in different diseases, no success or progress has been achieved in this area, which is possibly due to the fact that some diseases share similar pathways or mechanisms of action where MMPs and TIMPs intervene and they do not play the exact same role. Common processes among the different diseases mentioned include inflammation, angiogenesis, cell death, and migration, among others. Our understanding of the subtleties of MMP biology is inadequate, so it is necessary to consider key points to increase knowledge in this field of study.
References
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73.
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28–35.
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183.
- Fallata, A.M.; Wyatt, R.A.; Levesque, J.M.; Dufour, A.; Overall, C.M.; Crawford, B.D. Intracellular Localization in Zebrafish Muscle and Conserved Sequence Features Suggest Roles for Gelatinase A Moonlighting in Sarcomere Maintenance. Biomedicines 2019, 7, 93.
- Ali, M.A.; Chow, A.K.; Kandasamy, A.D.; Fan, X.; West, L.J.; Crawford, B.D.; Simmen, T.; Schulz, R. Mechanisms of cytosolic targeting of matrix metalloproteinase-2. J. Cell. Physiol. 2012, 227, 3397–3404.
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53.
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254.
- Wang, X.; Khalil, R.A. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharm. 2018, 81, 241–330.
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927.


