 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bartłomiej Surpeta | + 690 word(s) | 690 | 2020-04-16 09:32:10 | | | |
| 2 | Bruce Ren | Meta information modification | 690 | 2020-07-23 11:32:21 | | | | |
| 3 | Camila Xu | -26 word(s) | 664 | 2020-11-01 10:11:42 | | |
Video Upload Options
Protein dynamics is a highly complex phenomenon comprising numerous contributions from motions with different mechanisms of action and happening with diverse timescales and amplitudes that highly depend on the system and the local environment.
1. Introduction
Proteins are known to be dynamical entities, performing their function as an ensemble of diverse conformations rather than a single static structure. Protein dynamics is a highly complex phenomenon comprising numerous contributions from motions with different mechanisms of action and happening with diverse timescales and amplitudes (Figure 1) that highly depend on the system and the local environment. Subangstrom vibrations of covalent bonds represent the fastest of those movements. The exploration of various rotamers of side-chains and fluctuations of the protein backbone involve nontrivial moves that span the space of several angstroms. In protein cores, such moves can require several nanoseconds to execute due to the necessity to synchronize with changes in surrounding residues[3][4][5]. Many conformational changes involve slower and more prominent coordinated movements of several residues in a sequence that manifests as, for example, gating movement executed by loops surrounding the active sites of many proteins[6]. In ligand binding and unbinding events, especially when the binding site is deeply buried in the protein structure, ligands often have to travel tens of angstroms. Such a transport process requires a series of systematic adjustments of protein side-chains and backbones along the traversed paths that might take up hundreds of milliseconds to occur[7]. Among the slowest principal motions performed by proteins are highly organized collective translocations of whole domains, starting on microsecond timescales and with amplitudes reaching nanometers. Finally, the most extensive conformational change transpires during the protein (un)folding processes, which can take hours and even days[8][9][10].
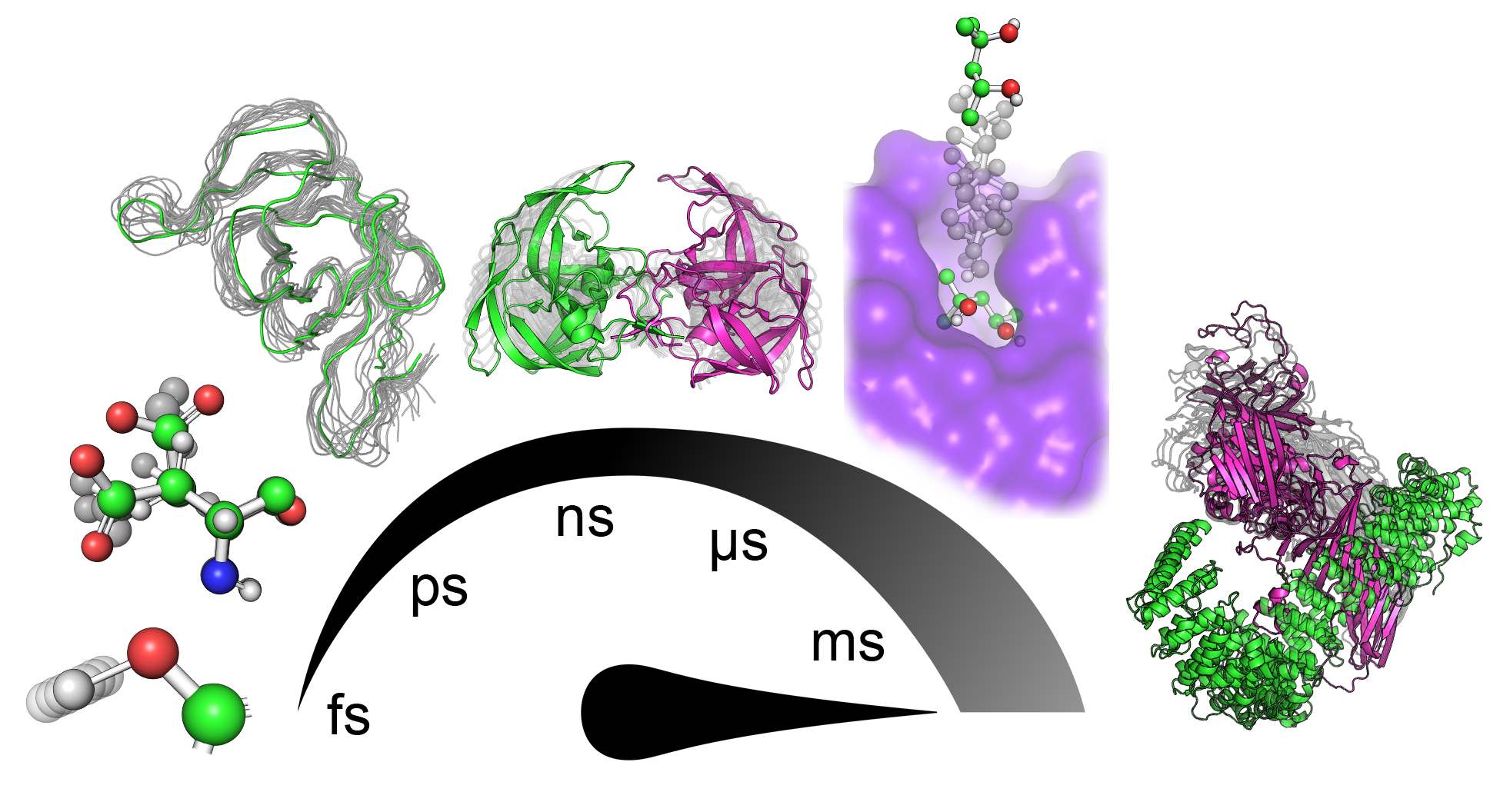
Figure 1. Hierarchy of principal motions in protein dynamics. From left to right: bond vibrations (fs–ps), side-chain rotations (ps–ns), backbone fluctuations (ns), loop motion/gating (ns–ms), ligand binding/unbinding events (>100 ns), and collective domain movement (>µs).
At present, continuous development of experimental techniques enables investigation of protein dynamics by methods such as: solution nuclear magnetic resonance (NMR) capable of capturing protein motions on broad timescales, fluorescence spectroscopy, or time-resolved macromolecular crystallography (X-ray)[8][9]. Furthermore, due to availability of enormous amount of high-quality three-dimensional (3D) protein structures obtained by NMR, X-ray, cryogenic electron microscopy (Cryo-EM), homology modeling, or hybrid-approaches, computational biophysical methods are progressively more applied to study protein motions at the atomic resolution, the most prominent being molecular dynamics (MD) simulation and normal mode analyses (NMA)[10][13].
When we consider the reliable simulation of protein dynamics as an essential component, it is natural to resort to the MD simulation technique as a golden standard to investigate the conformational behavior of a protein. Nowadays, various MD simulation protocols can be utilized to deliver insights into protein dynamics on millisecond timescales with the growing utilization of graphics processing unit (GPU)-enabled parallelism, development of more efficient software, enhanced sampling methods (high-throughput MD, Gaussian accelerated MD, etc.) and approximated models (e.g. coarse-graining), gradually making such simulations even more affordable[14][15][16][17][18][19][20][21].
2. Discussion
Despite all these improvements, MD simulations are not without errors in reproducing a realistic protein ensemble and, hence their experimental confirmation is necessary. Among the major limitations is the accuracy of force fields used to calculate interatomic interactions and the tractable sampling of the ensemble discussed above. The quality of traditionally applied force fields is intrinsically limited by numerous approximations like the lack of particular interaction types[22], neglect of electronic polarizability[23], and fixed protonation states of titrable residues[24]. At the expense of increased computational demands, some of those limitations can be partially overcome by improving potential models[25], resorting to polarizable force fields[26], and constant pH simulations[27]. Nonetheless, even without these advances, MD simulations relying on the latest force fields have been shown to reach chemical accuracy in their predictions for many different scenarios[28][29][30]. It is worth noting, including protein dynamics is crucial for design and re-design tasks, and as a recent trend, was described in recent review[31].
References
- H Frauenfelder; S. Sligar; P. Wolynes; The energy landscapes and motions of proteins. Science 1991, 254, 1598-1603, 10.1126/science.1749933.
- Pratul K Agarwal; Enzymes: An integrated view of structure, dynamics and function. Microbial Cell Factories 2006, 5, 2-2, 10.1186/1475-2859-5-2.
- Shimon Weiss; Fluorescence Spectroscopy of Single Biomolecules. Science 1999, 283, 1676-1683, 10.1126/science.283.5408.1676.
- Keith Moffat; The frontiers of time-resolved macromolecular crystallography: movies and chirped X-ray pulses.. Faraday Discussions 2003, 122, 65-77, 10.1039/b201620f.
- F. Schotte; Barry Sinervo; Jean Clobert; Watching a Protein as it Functions with 150-ps Time-Resolved X-ray Crystallography. Science 2003, 300, 1944-1947, 10.1126/science.1078797.
- Scott A. Hollingsworth; Ron O. Dror; Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129-1143, 10.1016/j.neuron.2018.08.011.
- Hiroshi Wako; Shigeru Endo; Normal mode analysis as a method to derive protein dynamics information from the Protein Data Bank. Biophysical Reviews 2017, 9, 877-893, 10.1007/s12551-017-0330-2.
- Katherine Henzler-Wildman; Rothee Kern; Dynamic personalities of proteins. Nature 2007, 450, 964-972, 10.1038/nature06522.
- Zoltán Gáspári; András Perczel; Protein Dynamics as Reported by NMR. Annual Reports on NMR Spectroscopy 2010, 71, 35-75, 10.1016/b978-0-08-089054-8.00002-2.
- Józef R. Lewandowski; Meghan E. Halse; M. Blackledge; Lyndon Emsley; Direct observation of hierarchical protein dynamics. Science 2015, 348, 578-581, 10.1126/science.aaa6111.
- James Kempf; J. Patrick Loria; Protein Dynamics from Solution NMR. Cell Biophysics 2002, 37, 187-212, 10.1385/cbb:37:3:187.
- Shimon Weiss; Fluorescence Spectroscopy of Single Biomolecules. Science 1999, 283, 1676-1683, 10.1126/science.283.5408.1676.
- Artur Góra; Jan Brezovsky; Jiří Damborský; Gates of Enzymes. Chemical Reviews 2013, 113, 5871-5923, 10.1021/cr300384w.
- Levi C.T. Pierce; Romelia Salomon-Ferrer; Cesar Augusto F. De Oliveira; J. Andrew McCammon; Ross C. Walker; Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. Journal of Chemical Theory and Computation 2012, 8, 2997-3002, 10.1021/ct300284c.
- Frank Noe; Beating the millisecond barrier in molecular dynamics simulations.. Biophysical Journal 2015, 108, 228-9, 10.1016/j.bpj.2014.11.3477.
- Mohammad Sultan; Rajiah Aldrin Denny; Ray Unwalla; Frank Lovering; Vijay S. Pande; Millisecond dynamics of BTK reveal kinome-wide conformational plasticity within the apo kinase domain. Scientific Reports 2017, 7, 15604, 10.1038/s41598-017-10697-0.
- Daniel-Adriano Silva; Dahlia R. Weiss; Fátima Pardo Avila; Lin-Tai Da; Michael Levitt; Dong Wang; Xuhui Huang; Millisecond dynamics of RNA polymerase II translocation at atomic resolution. Proceedings of the National Academy of Sciences 2014, 111, 7665-7670, 10.1073/pnas.1315751111.
- Romelia Salomon-Ferrer; Andreas W. Götz; Duncan Poole; Scott Le Grand; Ross C. Walker; Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of Chemical Theory and Computation 2013, 9, 3878-3888, 10.1021/ct400314y.
- Stefan Doerr; M. J. Harvey; Frank Noe; Gianni De Fabritiis; HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. Journal of Chemical Theory and Computation 2016, 12, 1845-1852, 10.1021/acs.jctc.6b00049.
- Yinglong Miao; Victoria A. Feher; J. Andrew McCammon; Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. Journal of Chemical Theory and Computation 2015, 11, 3584-3595, 10.1021/acs.jctc.5b00436.
- Matias R. Machado; Ari Zeida; Leonardo Darré; Sergio Pantano; From quantum to subcellular scales: multi-scale simulation approaches and the SIRAH force field. Interface Focus 2019, 9, 20180085, 10.1098/rsfs.2018.0085.
- Pengfei Li; Kenneth M. Merz; Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. Journal of Chemical Theory and Computation 2013, 10, 289-297, 10.1021/ct400751u.
- Zhifeng Jing; Chengwen Liu; Sara Y. Cheng; Rui Qi; Brandon D. Walker; Jean-Philip Piquemal; Pengyu Ren; Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications.. Annual Review of Biophysics 2019, 48, 371-394, 10.1146/annurev-biophys-070317-033349.
- John Mongan; David A Case; Biomolecular simulations at constant pH. Current Opinion in Structural Biology 2005, 15, 157-163, 10.1016/j.sbi.2005.02.002.
- Maria T. Panteva; George Giambasu; Darrin M. York; Comparison of structural, thermodynamic, kinetic and mass transport properties of Mg(2+) ion models commonly used in biomolecular simulations.. Journal of Computational Chemistry 2015, 36, 970-82, 10.1002/jcc.23881.
- Anhui Wang; Zhichao Zhang; Guohui Li; Higher Accuracy Achieved in the Simulations of Protein Structure Refinement, Protein Folding, and Intrinsically Disordered Proteins Using Polarizable Force Fields. The Journal of Physical Chemistry Letters 2018, 9, 7110-7116, 10.1021/acs.jpclett.8b03471.
- Plamen Dobrev; Sahithya Phani Babu Vemulapalli; Nilamoni Nath; Christian Griesinger; Helmut Grubmüller; Probing the Accuracy of Explicit Solvent Constant pH Molecular Dynamics Simulations for Peptides. Journal of Chemical Theory and Computation 2020, 16, 2561-2569, 10.1021/acs.jctc.9b01232.
- Louis G. Smith; Zhen Tan; Aleksandar Spasic; Debapratim Dutta; Leslie A. Salas-Estrada; Alan Grossfield; David H. Mathews; Chemically Accurate Relative Folding Stability of RNA Hairpins from Molecular Simulations. Journal of Chemical Theory and Computation 2018, 14, 6598-6612, 10.1021/acs.jctc.8b00633.
- Lim Heo; Michael Feig; Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proceedings of the National Academy of Sciences 2018, 115, 13276-13281, 10.1073/pnas.1811364115.
- Chuan Tian; Koushik Kasavajhala; Kellon A. A. Belfon; Lauren Raguette; He Huang; Angela N. Migues; John Bickel; Yuzhang Wang; Jorge Pincay; Qin Wu; et al.Carlos Simmerling ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. Journal of Chemical Theory and Computation 2019, 16, 528-552, 10.1021/acs.jctc.9b00591.
- Bartłomiej Surpeta; Carlos Eduardo Sequeiros-Borja; Jan Brezovsky; Dynamics, a Powerful Component of Current and Future in Silico Approaches for Protein Design and Engineering.. International Journal of Molecular Sciences 2020, 21, 2713, 10.3390/ijms21082713.

