 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elvira Escribano | + 2074 word(s) | 2074 | 2020-11-24 10:11:30 |
Video Upload Options
Blood contains various plasma proteins and cells to which endogenous and exogenous molecules can bind together to be transported throughout the circulatory system. Among the different plasma proteins, the binding of small molecular weight drug molecules is mostly associated with serum albumin and alpha-1 acid glycoprotein and, to a lesser extent globulins and lipoproteins. Although linear binding applies for most clinically used drugs, some physiopathological factors and/or dietary-drug interactions may lead to alterations of the drug-protein binding, which in turn may result in clinically changes in the pharmacokinetics and pharmacodynamics of the drug. As polyphenols (flavonoids and non-flavonoids) are widely present in plant-derived feeds, beberages, herbal medicines, and dietary supplements, the knowledge of how they bind to plasma proteins can prevent/avoid such interactions.
1. Introduction
The biological activities of dietary phenolic substances, present in plant-derived feeds, foods, beverages, herbal medicines, and dietary supplements, are of great interest. Polyphenols, which can be classified as flavonoids and non-flavonoids, contain, in addition to other substituents, at least one aromatic ring with one or more hydroxyl groups [1][2]. Flavonoids are a group of natural substances with variable phenolic structures as flavonols, flavan-3-ols (monomeric and polymeric structures), flavones, isoflavones, flavanones, and anthocyanidins. On the other hand, stilbenes, hydrolyzable tannins, lignans, and phenolic acids can be classified as non-flavonoids [3].
The absorption, distribution and elimination of dietary phytochemicals depend on their intestinal permeability and the influence of pre-systemic enzymes and/or transporters [4]. As systemic exposure can reflect tissue exposure, greater bioavailability should result in higher levels in tissues. Bioavailability is defined as the rate and the extent to which the active ingredient/phytochemical or moiety is absorbed from the ingested matrix and becomes available at the site of action [5]. It is well known that phenolic compounds have a low oral bioavailability, and undergo an extensive biotransformation mediated by phase I and phase II reactions in enterocytes and the liver, as well as by gut microbiota[6]. Polyphenol metabolites are also attracting research interest as their biological effects can be similar to or greater than those of the parent compounds [6][7]. Paradoxically, despite low oral bioavailability, most of the phenolic compounds have proven to have significant biological effects [6].
Once a xenobiotic has entered the systemic circulation, its rate of distribution to the various tissues of the body will be influenced by tissue hemodynamics (blood flow) and the ease with which it can cross the lipoidal cell membranes, either by passive diffusion or by passive/active facilitated transport (carrier-mediated) [8]. Nevertheless, the extent of distribution depends on partitioning into fat and other tissues and on unspecific/specific reversible binding to plasma proteins [9]. Plasma proteins, also called serum proteins, constitute important organic components consisting of simple as well as conjugated proteins [10]. Drugs are transported in the circulation either in a free form, dissolved in the aqueous phase of plasma, or in complex bonds with plasma proteins [11] in varying degrees [12]. Following the principle of reversible equilibrium and the law of mass action [13], an equilibrium exists between bound and free (unbound) molecular forms—additionally because binding is generally reversible [12]. Only the free form is capable of diffusing through membranes and from the vascular space into tissues, being eliminated by metabolism or excretion [14], and therefore pharmacological activity is exerted by the free drug concentration [15][16]. The fraction of a xenobiotic bound to a plasma protein depends on protein affinity towards the compound, protein and compound molar concentration, as well as on the possible competition with other endogenous and exogenous compounds for binding sites [17]. Generally, acidic compounds tend to bind to albumin, basic compounds to α1-acid glycoprotein (AAG), neutral compounds can be bound to both human serum albumin (HSA) and AAG, and neutral lipophilic compounds to lipoproteins. Other proteins, such as α-globulin, transcortin, fibrinogen, sex-hormone-binding globulin, and thyroid-binding globulin, bind specific compounds [18][19].
The free drug/xenobiotic concentration depends on the unbound drug clearance and dose, and is not usually changed by plasma protein binding (PPB) [16]. At a steady state, the free drug concentration remains balanced on both sides of any biomembrane [16]. As drug clearance occurs, a new equilibrium between bound and unbound forms is reached, which acts to maintain the free drug fraction [14]. Drug–protein complexes in plasma also serve as a drug reservoir, replacing what is removed by various distribution and elimination processes [8][15][17][20].
The free drug fraction is the ratio between the free drug concentration and total drug plasma concentration, which has values between zero (totally bound) and one (totally free), and remains constant in normal physiological conditions and at low molar drug concentrations. Only if the free fraction remains constant can the total plasma concentration be considered a good measure of changes in the unbound drug/xenobiotic concentration. This concept is important because the total plasma concentration is what is usually measured and not the unbound concentration, which is only occasionally determined. Although at the therapeutic concentration of most drugs, the molar concentration of unbound drugs is usually low in comparison with the molar concentration of the protein binding sites [13][21], in some pathophysiological conditions, the free drug fraction can be reduced/increased with ensuing changes in the distribution process, either by an altered protein–drug affinity or by a change in plasma protein levels [16].
Although it is traditionally considered that only the free form is capable of diffusing through membranes, recent studies have hypothesized that HSA facilitates Dp44mT delivery to the lysosomes by internalization through a carrier mediated transport mechanism enhancing its anti-cancer activity [22]. In addition, an in vitro study has shown for proteins with a net negative charge such as albumin, and for drugs highly bound to albumin and in the physiological albumin concentration range, a protein-mediated uptake mechanism [23]. It was observed in hepatocytes and cardiac myocytes. Certainly, this requires further intense investigation since PPB is an important process that determines the pharmacological activity and pharmacokinetics of drugs and other xenobiotics [12][24], and the impact of PPB on the efficacy and safety of a treatment needs to be better understood [25].
As with other xenobiotics, the distribution of phenolic compounds to the various tissues of the body is influenced by unspecific, reversible binding events to plasma protein [9]. PPB and phenolics have been the subject of numerous recent studies, which have focused above all on structure–affinity relationships. The aim of this review is to summarize how structural modifications affect the affinity of the main dietary polyphenols and their metabolites for HSA and to elucidate the main factors involved in their binding and the binding site. Drug binding properties of HSA and competitive binding with the most widespread dietary phenolic compounds are also covered.
2. Plasma Proteins
Plasma contains various proteins with different functions including the transport throughout the circulatory system of endogenous and exogenous molecules. The binding of small molecular weight drug molecules with plasma proteins is mostly associated with HSA and AAG and, to a lesser extent, globulins and lipoproteins [26].
HSA is a 66 kDa non-glycosylated monomeric protein of 585 amino acids present in the human body at a concentration of 0.53–0.75 mM [20][27]. HSA constitutes ~4.5% of the weight of human blood and helps to maintain osmotic pressure and pH in the blood stream [26][27]. Its principal functions are to transport fatty acids, hormones, drugs, nutrients and inorganic ions and to buffer pH [28][29]. Due to a large content of ionic residues, HSA is highly soluble in water and its flexibility allows specific binding to a wide array of molecules [26]. The polypeptide chain of HSA forms a heart-shaped conformation with approximate dimensions of 80 × 80 × 30 Å, about 67% consisting of α-helices [11][28]. It contains three homologous α-helical domains (I–III), each further divided into two subdomains (A and B) [30]. Among them, subdomains IIA and IIIA are two important binding sites. They are delimited by a hydrophobic surface on one side and a positively charged surface on the other side, displaying well determined cavities to specifically bind neutral and negatively charged compounds.
The globulins (α-globulins, β-globulins, and γ-globulin) are a group of globular water-insoluble proteins [31]. AAG, also known as orosomucoid, is acidic, heavily glycosylated (38 to 48 kDa protein, concentration ~12–31 µM), and comprises a single amino acid chain of 204 residues. An acute phase plasma protein, it is the principal extracellular lipocalin present in blood [32]. Multiple drug-binding sites have been reported for AAG, but one appears to be most important, particularly for basic and neutral drugs [8][27]. It should also be considered that if a compound is available as a racemic mixture in blood/plasma, both HSA and AAG are able to bind preferentially to one stereoisomer [25].
To date, two different approaches to assess drug–protein binding can be distinguished. On one hand, separative methods used to determine directly either the unbound drug or the bound drug concentration by separation of the free ligand from the bound species can be classified as conventional methods (equilibrium dialysis, ultrafiltration and ultracentrifugation), high-performance affinity chromatography and capillary electrophoresis–frontal analysis. On the other hand, non-separative methods have been developed to characterize drug–protein interactions. In this sense, spectroscopic methods (UV–visible, fluorescence, infrared, nuclear magnetic resonance, optical rotatory dispersion, and circular dichroism) based on the perturbation of electronic and spectroscopic energy levels of the ligand or the protein and calorimetric techniques (isothermal titration calorimetry and differential scanning calorimetry) have been extensively used. In the last year, computational measurements have also been developed to characterize the polyphenol–protein interactions [1][24].
3. Phenolic Compounds–Drug Interaction
Data on food–drug interactions are generally scarce, despite some well documented exceptions (e.g., grapefruit juice and statins), as food consumption and herbal teas/beverages are not usually monitored in patients. Interactions occur after the concomitant intake of food and drugs, with impacts on the absorption and/or metabolism of the active substance. In some cases, the effects of the interactions may benefit the patients, but they frequently undermine the efficacy of the drug or induce adverse reactions [33].
In the case of PPB, a hormone, drug or even a toxin can be displaced by competing phenolic compounds and then circulates in the blood in a free form. The pharmacodynamics and pharmacokinetics of a drug may subsequently be modified, potentially leading to stronger pharmacological activity, adverse effects and faster elimination [34]. It should be noted that such effects are rarely caused by the formation of phenol–HSA complexes, as the low phenol and high HSA concentration in plasma renders saturation at the binding site unlikely. Moreover, phenolic compounds are often subject to high first-pass metabolism, and thus it is the conjugated-HSA complex that should be taken into account in a potential food–drug interaction. Nevertheless, such an outcome should be kept in mind for drugs with high PPB, high hepatic/renal extraction ratio and narrow therapeutic index, and for other plasma proteins more specific than HSA.
To date, most of the research on food–drug interactions has been focused on flavonoids and HSA. Rutin and baicalin have been extensively used to determine the effect of flavonoids on the binding properties of cleviprex, theophylline, nifedipine, promethazine and ticagrelor [33][35][36][37][38]. The results show that (1) both hydrogen bonds and hydrophobic interactions play a central role in the binding process, which is spontaneous; (2) flavonoids can reduce the association constant and increase the distance of drugs binding to HSA due to competitive binding at site I; (3) the synergistic effect of drugs with rutin and baicalin can further change the HSA conformation, and (4) reduced affinities of drugs binding to HSA in the presence of flavonoids may lead to an increase in free drugs in the blood, which would affect their transportation and/or disposition and may provoke adverse or toxic effects, as shown in Figure 1.
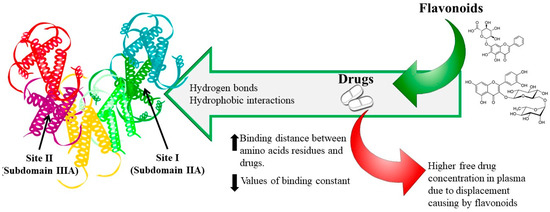
Figure 1. Dietary flavonoid–drug interaction mechanism.
Quercetin had the same effect as rutin and baicalin on ticagrelor and propranolol binding to HSA [38][39]. In 2012, Maciazek-Jurczyk and collaborators reported that competition from curcumin for the binding site of tamoxifen in HSA reduced the binding affinity of this chemopreventive agent, which increased its unbound fraction in the blood with potentially toxic effects [40]. In 2017, Rimac and colleagues showed that warfarin–flavonoid interactions should be regarded as negligible, as they do not share the same binding region in HSA [41]. Conversely, in the same year, it was demonstrated that quercetin metabolites strongly displace warfarin when binding to HSA, suggesting that high quercetin levels can negatively interfere with warfarin therapy [42].
Consequently, the intake of flavonoid-rich foods and beverages should be reduced during treatment with the aforementioned drugs to avoid food–drug interactions and the incidence of toxic symptoms. Alternatively, drugs that do not share the same binding region as flavonoids can be used.
References
- Xiao, J.; Kai, G. A Review of Dietary Polyphenol-Plasma Protein Interactions: Characterization, Influence on the Bioactivity, and Structure-Affinity Relationship. Crit. Rev. Food Sci. Nutr. 2012, 52, 85–101.
- Ribas-Agustí, A.; Martín-Belloso, O.; Soliva-Fortuny, R.; Elez-Martínez, P. Food processing strategies to enhance phenolic compounds bioaccessibility and bioavailability in plant-based foods. Crit. Rev. Food Sci. Nutr. 2018, 58, 2531–2548.
- Poloni, D.M.; Dangles, O.; Vinson, J.A. Binding of Plant Polyphenols to Serum Albumin and LDL: Healthy Implications for Heart Disease. J. Agric. Food Chem. 2019, 67, 9139–9147.
- FDA, G. Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General. 2002.
- Neilson, A.P.; Goodrich, K.M.; Ferruzzi, M.G. Bioavailability and Metabolism of Bioactive Compounds From Foods. In Nutrition in the Prevention and Treatment of Disease; Elsevier, 2017; pp. 301–319 ISBN 978-0-12-802928-2.
- Luca, S.V.; Macovei, I.; Bujor, A.; Miron, A.; Skalicka-Woźniak, K.; Aprotosoaie, A.C.; Trifan, A. Bioactivity of dietary polyphenols: The role of metabolites. Crit. Rev. Food Sci. Nutr. 2020, 60, 626–659.
- Serreli, G.; Deiana, M. Biological Relevance of Extra Virgin Olive Oil Polyphenols Metabolites. Antioxidants 2018, 7, 170.
- Caldwell, J.; Gardner, I.; Swales, N. An Introduction to Drug Disposition: The Basic Principles of Absorption, Distribution, Metabolism, and Excretion. Toxicol. Pathol. 1995, 23, 102–114.
- ADME Processes in Pharmaceutical Sciences: Dosage, Design, and Pharmacotherapy Success; Talevi, A., Quiroga, P.A.M., Eds.; Springer International Publishing: Cham, 2018; ISBN 978-3-319-99592-2.
- Gupta, A. Plasma Proteins. In Comprehensive Biochemistry for Dentistry: Textbook for Dental Students; Gupta, A., Ed.; Springer Singapore: Singapore, 2019; pp. 67–75 ISBN 978-981-13-1035-5.
- Otagiri, M. A Molecular Functional Study on the Interactions of Drugs with Plasma Proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323.
- Kamble, S.; Loadman, P.; Abraham, M.H.; Liu, X. Structural properties governing drug-plasma protein binding determined by high-performance liquid chromatography method. J. Pharm. Biomed. Anal. 2018, 149, 16–21.
- Dasgupta, A. Monitoring Free Drug Concentration. In Clinical Challenges in Therapeutic Drug Monitoring; Elsevier, 2016; pp. 71–100 ISBN 978-0-12-802025-8.
- Roberts, J.A.; Pea, F.; Lipman, J. The Clinical Relevance of Plasma Protein Binding Changes. Clin. Pharmacokinet. 2013, 52, 1–8.
- Vallner, J.J. Binding of Drugs by Albumin Plasma Protein. J. Pharm. Sci. 1977, 66, 447–465.
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939.
- Beasley, V. Absorption, Distribution, Metabolism, and Elimination: Differences Among Species. Vet. Toxicol. 1999, 1–19.
- Hartmut, D.; Schmidt, S. Clinical pharmacokinetics and pharmacodynamics: concepts and applications; 5th ed.; Wolters Kluwer Health/Lippincott William & Wilkins: Philadelphia, Pa., 2020;
- Rowland, M.; Tozer, T.N. Clinical pharmacokinetics and pharmacodynamics: concepts and applications; 4th ed.; Wolters Kluwer Health/Lippincott William & Wilkins: Philadelphia, Pa., 2011; ISBN 978-0-7817-5009-7.
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: a new approach. Biochem. Pharmacol. 2002, 64, 1355–1374.
- Nation, R.L. Concentration-dependent plasma protein binding: Expect the unexpected. Eur. J. Pharm. Sci. 2018, 6.
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—more than just a serum protein. Front. Physiol. 2014, 5, 299.
- Poulin, P.; Haddad, S. Albumin and Uptake of Drugs in Cells: Additional Validation Exercises of a Recently Published Equation that Quantifies the Albumin-Facilitated Uptake Mechanism(s) in Physiologically Based Pharmacokinetic and Pharmacodynamic Modeling Research. J. Pharm. Sci. 2015, 104, 4448–4458.
- Vuignier, K.; Schappler, J.; Veuthey, J.-L.; Carrupt, P.-A.; Martel, S. Drug–protein binding: a critical review of analytical tools. Anal. Bioanal. Chem. 2010, 398, 53–66.
- Schmidt, S.; Gonzalez, D.; Derendorf, H. Significance of Protein Binding in Pharmacokinetics and Pharmacodynamics. J. Pharm. Sci. 2010, 99, 1107–1122.
- Howard, M.; Hill, J.; Galluppi, G.; McLean, M. Plasma Protein Binding in Drug Discovery and Development. Comb. Chem. High Throughput Screen. 2010, 13, 170–187.
- Trainor, G.L. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discov. 2007, 2, 51–64.
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835.
- Sheng, F.; Wang, Y.; Zhao, X.; Tian, N.; Hu, H.; Li, P. Separation and Identification of Anthocyanin Extracted from Mulberry Fruit and the Pigment Binding Properties toward Human Serum Albumin. J. Agric. Food Chem. 2014, 62, 6813–6819.
- Varshney, A.; Sen, P.; Ahmad, E.; Rehan, Mohd.; Subbarao, N.; Khan, R.H. Ligand binding strategies of human serum albumin: How can the cargo be utilized? Chirality 2010, 22, 77–87.
- Gupta, A. Comprehensive Biochemistry for Dentistry: Textbook for Dental Students.; Springer: Singapore, 2018; ISBN 9789811310348.
- Yeggoni, D.P.; Rachamallu, A.; Subramanyam, R. A comparative binding mechanism between human serum albumin and α-1-acid glycoprotein with corilagin: biophysical and computational approach. RSC Adv. 2016, 6, 40225–40237.
- Gokara, M.; Sudhamalla, B.; Amooru, D.G.; Subramanyam, R. Molecular Interaction Studies of Trimethoxy Flavone with Human Serum Albumin. PLoS ONE 2010, 5, e8834, doi:10.1371/journal.pone.0008834.
- Boulton, D.W.; Walle, U.K.; Walle, T. Extensive Binding of the Bioflavonoid Quercetin to Human Plasma Proteins. J. Pharm. Pharmacol. 1998, 50, 243–249.
- Nozaki, A.; Kimura, T.; Ito, H.; Hatano, T. Interaction of Polyphenolic Metabolites with Human Serum Albumin: A Circular Dichroism Study. Chem. Pharm. Bull. (Tokyo) 2009, 57, 1019–1023.
- Wang, X.; Guo, X.-Y.; Xu, L.; Liu, B.; Zhou, L.-L.; Wang, X.-F.; Wang, D.; Sun, T. Studies on thecompetitive binding of cleviprex and flavonoids to plasma protein by multi-spectroscopic methods: A prediction of food-drug interaction. J. Photochem. Photobiol. B 2017, 175, 192–199.
- Wang, X.; He, L.-L.; Liu, B.; Wang, X.; Xu, L.; Wang, X.-F.; Sun, T. Decrease of the affinity of theophylline bind to serum proteins induced by flavonoids and their synergies on protein conformation. Int. J. Biol. Macromol. 2018, 107, 1066–1073.
- Wang, X.; Liu, Y.; He, L.-L.; Liu, B.; Zhang, S.-Y.; Ye, X.; Jing, J.-J.; Zhang, J.-F.; Gao, M.; Wang, X. Spectroscopic investigation on the food components–drug interaction: The influence of flavonoids on the affinity of nifedipine to human serum albumin. Food Chem. Toxicol. 2015, 78, 42–51.
- He; Wang, Z.-X.; Wang, Y.-X.; Liu, X.-P.; Yang, Y.-J.; Gao, Y.-P.; Wang, X.; Liu, B.; Wang, X. Studies on the interaction between promethazine and human serum albumin in the presence of flavonoids by spectroscopic and molecular modeling techniques. Colloids Surf. B Biointerfaces 2016, 145, 820–829.
- Liu; Zhang, J.; Bai, C.-L.; Wang, X.; Qiu, X.-Z.; Wang, X.-L.; Ji, H.; Liu, B. Spectroscopic study on flavonoid–drug interactions: Competitive binding for human serum albumin between three flavonoid compounds and ticagrelor, a new antiplatelet drug. J. Lumin. 2015, 168, 69–76.
- Mohseni-Shahri, F.S.; Housaindokht, M.R.; Bozorgmehr, M.R.; Moosavi-Movahedi, A.A. The influence of the flavonoid quercetin on the interaction of propranolol with human serum albumin: Experimental and theoretical approaches. J. Lumin. 2014, 154, 229–240.
- Xiao, J.; Zhao, Y.; Wang, H.; Yuan, Y.; Yang, F.; Zhang, C.; Yamamoto, K. Noncovalent Interaction of Dietary Polyphenols with Common Human Plasma Proteins. J. Agric. Food Chem. 2011, 59, 10747–10754.


