 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christoph Rameshan | + 836 word(s) | 836 | 2020-04-15 12:17:24 | | | |
| 2 | Christoph Rameshan | Meta information modification | 836 | 2020-04-16 18:50:42 | | | | |
| 3 | Camila Xu | -1 word(s) | 835 | 2020-11-01 10:32:46 | | |
Video Upload Options
In heterogeneous catalysis, surfaces decorated with uniformly dispersed, catalytically-active (nano)particles are a key requirement for excellent performance. Besides standard catalyst preparation routines—with limitations in controlling catalyst surface structure (i.e., particle size distribution or dispersion)—exsolution is a potential novel and time efficient route to precisely tailor catalyst surface morphology and composition of perovskites:
Perovskite-type oxides of nominal composition ABO3 with transition metal cations on the B-site can exsolve the B-site transition metal upon controlled reduction. In this exsolution process, the transition metal emerges from the oxide lattice and migrates to the surface where it forms catalytically active nanoparticles. Doping the B-site with reducible and catalytically highly active elements, offers the opportunity of tailoring properties of exsolution catalysts.
1. Introduction
In heterogeneous catalysis, controllable surface properties and a maximum amount of stable uniformly-dispersed catalytically highly active sites on the surface of a porous material are of key importance. Typically, catalysts consist of metal, alloy, or oxide nanoparticles embedded in an oxide support material. Depending on the type of catalytic reaction, active sites are either just nanoparticles themselves or the combination of nanoparticles and oxide support. Ideally, catalysts exhibit long-term stability and activity during reaction. Therefore, resistance to catalyst poisons, inhibition of carbon deposition (blocking of active sites), and prevention of particle agglomeration and sintering (loss of active surface area) is essential.
Typically, these structures are prepared by deposition, impregnation or precipitation techniques[1][2] followed by catalyst activation prior to reactions via oxidation and reduction[3]. Although these approaches are applied widely, they offer limited control over size, distribution, and anchorage of deposited species, not only during preparation but also during catalyst activation or operation, and they might be time consuming and costly[4].
Alternatively, an emerging concept is to grow the nanoparticles in situ, by reduction or during catalytic reaction, directly from the (porous) oxide backbone support itself[4][5][6]. Perovskites (nominal composition ABO3 with A and B being a large and small cation, respectively, cf. Figure 1a) can incorporate catalytically highly active elements as cations on the B-site of the perovskite lattice (e.g., Ni[4][7], Fe [4][8], Co[9], Cu[4][9], Pt[10], Pd[10][11]). Upon exposure to reducing conditions, these elements can be partly exsolved as nanoparticles, thus opening the possibility of in situ growth of active catalysts[12], see sketch in Figure 1b. In comparison to traditional deposition techniques, this process produces finer and higher dispersed catalyst nanoparticles[4][13][14] and is more time- and cost-effective, as it does not require multiple "deposition" steps or expensive precursors[15]. Furthermore, it has been shown that nanoparticles formed by exsolution exhibit improved sintering stability during catalytic reaction[16]. In addition, due to a possible reversibility of the nanoparticle exsolution process, particle agglomeration can be avoided by reoxidation cycles[17], hence, greatly enhancing the catalyst lifetime[12][18].
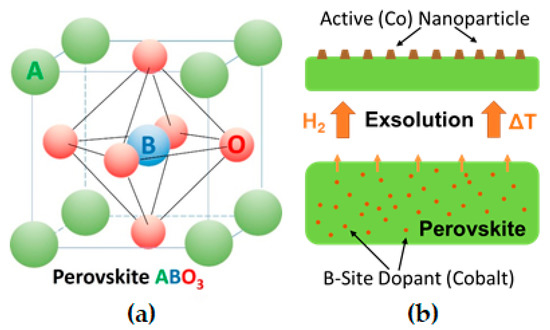
Mechanisms for nanoparticle exsolution from perovskites were investigated already by multiple groups[4][11][19][20][21]. It is generally accepted that exsolution catalysts need easily reducible dopant cations[22], such as Ru, Co, Pd, or Ni[23], that will form metallic deposits on the perovskite surface. Thus, depending on temperature and oxygen chemical potential in the gas phase (i.e., the strength of reduction), dopants and in some cases also lattice cations are reduced and exsolved as nanoparticles[8][24]. As an explanation for exsolution mechanisms, Neagu et al.[4] proposed that oxygen vacancies created by reduction destabilize the perovskite lattice, especially when their concentration is high and when additionally A-site cation vacancies are present. Such combined oxygen and A-site cation vacancy formations may locally cause spontaneous exsolution of B-site species in an attempt to re-establish stoichiometry across all sites. Oh et al.[19] implied that the morphological evolution of nanoparticles could be explained using a simple energy-based model, which accounts for interplay between surface free energy and strain energy induced by the included metal nucleate. By quantitative strain field modelling, they could demonstrate that this provides the driving force for exsolution processes. Similarly, Han and co-workers applied classical nucleation theory to the exsolution process and found the strain between metal particle and supporting oxide being a potential influencing factor for the particle size.
2. Conclusion
Usually, formation of nanoparticles on the surface is accomplished via a reductive treatment. For example, Arrivé et al.[25] showed for Ni doped LaSrTiO3 that pre-reduction in H2 at 1573 K leads to exsolution of Ni particles on the surface of solid oxide electrochemical cells leading to improved electrochemical performance. Additionally, Papargyriou and Irvine demonstrated Ni exsolution from Ni-doped La0.75Sr0.25Cr0.5Fe0.5O3 perovskite by H2 reduction[14]. Nanoparticles with a size of 30–50 nm were formed on the surface, leading to an improvement of catalytic properties of the material. Similarly, Sun et al.[13] could observe Ni nanoparticle exsolution from Ni-doped La0.7Sr0.3CrO3 upon pre-reduction in H2. Most of the recent studies on exsolution phenomena are reported and studied by the electrochemical community (mainly with respect to fuel cell technology), but utilization of this effect for preparing tailored heterogeneous catalyst materials is still rare—even though the initial idea came indeed from the catalysis community[12].
References
- R.J. Gorte; John M. Vohs; Nanostructured anodes for solid oxide fuel cells. Current Opinion in Colloid & Interface Science 2009, 14, 236-244, 10.1016/j.cocis.2009.04.006.
- John T. Yates; Charles T. Campbell; Surface chemistry: Key to control and advance myriad technologies. Proceedings of the National Academy of Sciences 2011, 108, 911-916, 10.1073/pnas.1006671107.
- Séverine Rousseau; Olivier Marie; Philippe Bazin; Marco Daturi; Stéphane Verdier; Virginie Harlé; Investigation of Methanol Oxidation over Au/Catalysts Using Operando IR Spectroscopy: Determination of the Active Sites, Intermediate/Spectator Species, and Reaction Mechanism. Journal of the American Chemical Society 2010, 132, 10832-10841, 10.1021/ja1028809.
- Dragos Neagu; George Tsekouras; David N. Miller; Hervé Ménard; John T. S. Irvine; In situ growth of nanoparticles through control of non-stoichiometry. Nature Chemistry 2013, 5, 916-923, 10.1038/nchem.1773.
- Michael B. Katz; Shuyi Zhang; Yingwen Duan; Hongjie Wang; Minghao Fang; Kui Zhang; Baihai Li; George W. Graham; X. Q. Pan; Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: Bulk versus surface re-dispersion. Journal of Catalysis 2012, 293, 145-148, 10.1016/j.jcat.2012.06.017.
- George Tsekouras; Dragos Neagu; John T. S. Irvine; Step-change in high temperature steam electrolysis performance of perovskite oxide cathodes with exsolution of B-site dopants. Energy & Environmental Science 2013, 6, 256-266, 10.1039/c2ee22547f.
- W. Kobsiriphat; B. D. Madsen; Y. Wang; M. Shah; L. D. Marks; Scott A. Barnett; Nickel- and Ruthenium-Doped Lanthanum Chromite Anodes: Effects of Nanoscale Metal Precipitation on Solid Oxide Fuel Cell Performance. Journal of the Electrochemical Society 2010, 157, B279, 10.1149/1.3269993.
- Alexander Karl Opitz; Andreas Nenning; Christoph Rameshan; Raffael Rameshan; Raoul Blume; Michael Hävecker; Axel Knop-Gericke; Günther Rupprechter; Jürgen Fleig; Bernhard Klötzer; et al. Enhancing Electrochemical Water-Splitting Kinetics by Polarization-Driven Formation of Near-Surface Iron(0): An In Situ XPS Study on Perovskite-Type Electrodes**. Angewandte Chemie International Edition 2014, 54, 2628-2632, 10.1002/anie.201409527.
- Lawrence Adijanto; Venu Balaji Padmanabhan; Raymond J. Gorte; John M. Vohs; Polarization-Induced Hysteresis in CuCo-Doped Rare Earth Vanadates SOFC Anodes. Journal of the Electrochemical Society 2012, 159, F751-F756, 10.1149/2.042211jes.
- Hirohisa Tanaka; Mari Uenishi; Masashi Taniguchi; Isao Tan; Keiichi Narita; Mareo Kimura; Kimiyoshi Kaneko; Yasuo Nishihata; Jun’Ichiro Mizuki; The intelligent catalyst having the self-regenerative function of Pd, Rh and Pt for automotive emissions control. Catalysis Today 2006, 117, 321-328, 10.1016/j.cattod.2006.05.029.
- Michael B. Katz; George W. Graham; Yingwen Duan; Hong Liu; Carolina Adamo; Darrell G. Schlom; X. Q. Pan; Self-Regeneration of Pd–LaFeO3Catalysts: New Insight from Atomic-Resolution Electron Microscopy. Journal of the American Chemical Society 2011, 133, 18090-18093, 10.1021/ja2082284.
- Y. Nishihata; J. Mizuki; T. Akao; H. Tanaka; M. Uenishi; M. Kimura; T. Okamoto; N. Hamada; Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164-167, 10.1038/nature00893.
- Yifei Sun; Jianhui Li; Yimin Zeng; Babak Shalchi Amirkhiz; Mengni Wang; Yashar Behnamian; Jing-Li Luo; A-site deficient perovskite: the parent for in situ exsolution of highly active, regenerable nano-particles as SOFC anodes. Journal of Materials Chemistry A 2015, 3, 11048-11056, 10.1039/C5TA01733E.
- D. Papargyriou; John T. S. Irvine; Nickel nanocatalyst exsolution from (La,Sr) (Cr,M,Ni)O3 (M Mn,Fe) perovskites for the fuel oxidation layer of Oxygen Transport Membranes. Solid State Ionics 2016, 288, 120-123, 10.1016/j.ssi.2015.11.007.
- Qian Zhao; Harald Lorenz; Stuart Turner; Oleg Lebedev; Gustaaf Van Tendeloo; Christoph Rameshan; Bernhard Klötzer; Jürgen Konzett; Simon Penner; Catalytic characterization of pure SnO2 and GeO2 in methanol steam reforming. Applied Catalysis A: General 2010, 375, 188-195, 10.1016/j.apcata.2009.12.027.
- Dragos Neagu; Tae-Sik Oh; David N. Miller; Hervé Ménard; Syed M. Bukhari; Stephen R. Gamble; Raymond J. Gorte; John M. Vohs; John T. S. Irvine; Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nature Communications 2015, 6, 8120, 10.1038/ncomms9120.
- Dariusz Burnat; Roman Kontic; Lorenz Holzer; Patrick Steiger; Davide Ferri; Andre Heel; Smart material concept: reversible microstructural self-regeneration for catalytic applications. Journal of Materials Chemistry A 2016, 4, 11939-11948, 10.1039/C6TA03417A.
- Hirohisa Tanaka; Masashi Taniguchi; Mari Uenishi; Nobuhiko Kajita; Isao Tan; Yasuo Nishihata; Jun’Ichiro Mizuki; Keiichi Narita; Mareo Kimura; Kimiyoshi Kaneko; et al. Self-Regenerating Rh- and Pt-Based Perovskite Catalysts for Automotive-Emissions Control. Angewandte Chemie International Edition 2006, 45, 5998-6002, 10.1002/anie.200503938.
- Tae-Sik Oh; Ehsan K. Rahani; Dragos Neagu; John T. S. Irvine; Vivek B. Shenoy; Raymond J. Gorte; John M. Vohs; Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites during Exsolution. The Journal of Physical Chemistry Letters 2015, 6, 5106-5110, 10.1021/acs.jpclett.5b02292.
- Jacob M. Haag; Scott A. Barnett; James W. Richardson; Kenneth R. Poeppelmeier; Structural and Chemical Evolution of the SOFC Anode La0.30Sr0.70Fe0.70Cr0.30O3−δupon Reduction and Oxidation: An in Situ Neutron Diffraction Study. Chemistry of Materials 2010, 22, 3283-3289, 10.1021/cm100609e.
- Thomas Götsch; Lukas Schlicker; Maged F. Bekheet; Andrew Doran; Matthias Grünbacher; Corsin Praty; Mizuki Tada; Hirosuke Matsui; Nozomu Ishiguro; Aleksander Gurlo; et al.Bernhard KlötzerSimon Penner Structural investigations of La0.6Sr0.4FeO3−δ under reducing conditions: kinetic and thermodynamic limitations for phase transformations and iron exsolution phenomena. RSC Advances 2018, 8, 3120-3131, 10.1039/c7ra12309d.
- Ohhun Kwon; Sivaprakash Sengodan; Kyeounghak Kim; Gihyeon Kim; Hu Young Jeong; Jeeyoung Shin; Young-Wan Ju; Jeong Woo Han; Guntae Kim; Exsolution trends and co-segregation aspects of self-grown catalyst nanoparticles in perovskites. Nature Communications 2017, 8, 15967, 10.1038/ncomms15967.
- Hyeon Han; Jucheol Park; Sang Yeol Nam; Kun Joong Kim; Gyeong Man Choi; Stuart S. P. Parkin; Hyun Myung Jang; John T. S. Irvine; Lattice strain-enhanced exsolution of nanoparticles in thin films.. Nature Communications 2019, 10, 1471, 10.1038/s41467-019-09395-4.
- Thomas Götsch; Norbert Köpfle; Matthias Grünbacher; Johannes Bernardi; Emilia A. Carbonio; Michael Hävecker; Axel Knop-Gericke; Maged F. Bekheet; Lukas Schlicker; Andrew Doran; et al.Aleksander GurloAlexandra FranzBernhard KlötzerSimon PennerMichael HaeveckerBernhard Kloetzer Crystallographic and electronic evolution of lanthanum strontium ferrite (La0.6Sr0.4FeO3−δ) thin film and bulk model systems during iron exsolution. Physical Chemistry Chemical Physics 2019, 21, 3781-3794, 10.1039/c8cp07743f.
- Charline Arrivé; Thibaud Delahaye; Olivier Joubert; Gilles Gauthier; Exsolution of nickel nanoparticles at the surface of a conducting titanate as potential hydrogen electrode material for solid oxide electrochemical cells. Journal of Power Sources 2013, 223, 341-348, 10.1016/j.jpowsour.2012.09.062.

