 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jing Sun | + 4124 word(s) | 4124 | 2020-12-18 04:26:51 | | | |
| 2 | Nicole Yin | + 2 word(s) | 4126 | 2020-12-29 04:15:51 | | |
Video Upload Options
Thermoresponsive polypeptoids exhibiting a reversible phase transition in a controlled manner to temperature are a promising class of smart polymers that have drawn growing interest because of its excellent biocompatibility, biodegradability, and bioactivity. The phase transition behavior of these polymers can be tuned by polymer architectures, chain-end, and various functional groups.
1.Introduction
The polypeptoids are highly designable, where two fundamental synthetic approaches are applied: solid-phase submonomer synthesis and ring-opening polymerization (ROP) of N-substituted glycine N-carboxyanhydrides (NCA) monomer or N-substituted N-thiocarboxyanhydrides (NTA) monomer. The peptoid oligomers with short chain length, specific sequence, and monodispersity can be achieved by the solid-phase synthetic method. Such method enables precise control over the functional properties of the peptoids[1]. In contrast to the solid-phase method, the ROP approach offers an effective way to produce the polypeptoids with high molecular weights and large-scale yields[2][3][4]. In this case, two distinct methods for preparation of functional polypeptoids have been reported including the controlled polymerization of functional monomer and post-polymerization modification (PPM) of well-defined polypeptoid precursors[5]. The polymerization of functionalized monomer method allows for the efficient preparation of the polypeptoids with quantitative functionality and various functional monomers on one individual polymer chain. However, the synthesis of the monomers and controlled polymerization remains a challenge. In contrast, the post-polymerization modification that typically involves “click chemistry” significantly simplifies the synthetic routes, which provides an effective and convenient tool to prepare versatile polypeptoids. In this review, we will mainly discuss recent advances on the design and synthesis of thermoresponsive polypeptoids based on the ROP of NCA or NTA, as well as their biological applications.
2. Synthetic Strategies of Thermoresponsive Polypeptoids
Polypeptoids can be prepared by two fundamental synthetic methods including solid-phase submonomer synthesis and ROP of NCA or NTA. Zuckermann et al. developed a two-step submonomer synthetic method to synthesize peptoids: an acylation step with a haloacetic acid and a displacement reaction with a primary amine, which was an analogy to the well-established Merrifield method of solid-phase peptide synthesis (SPPS)[6][7]. With this approach, high synthesis efficiency and limited chain length, generally up to 50 monomers, have been obtained. In addition, the methods of ROP of NCA or NTA used to synthesize polypeptoids can achieve high molecular weights (MWs) of polypeptoids and large-scale yields[8][9]. The synthesis of polypeptoids can be accomplished by the ROP of NCA monomers using primary amine or N-heterocyclic carbene (NHC) as initiators. Zhang and coworkers reported NHC-mediated polymerization of NCA monomers, by which polymerization efficiency and MW can be well controlled in solutions with low dielectric constants[10][11]. In addition to N-substituted NCAs, ROP of NTA provides a new synthetic method to prepare polypeptoids and in recent years, Ling et al. prepared a series of homopolypeptoids and copolypeptoids with high MWs through controlled ROP of NTA monomers and further explored their physicochemical properties[9][12][13].
2.1. The Controlled Polymerization of Functional Monomer Strategy
The polymerization of functional monomer shows prominent advantages to produce the polypeptoids with quantitative modification. In addition, versatile functional monomers can be efficiently incorporated into one individual polymer chain. The polypeptoid monomers for ROP typically include NCA and NTA monomers. A couple of methods have been reported to prepare the monomers[14]. Particularly, NCA monomers can be synthesized from the N-substituted glycine precursors in three steps (Scheme 1)[15]. Although the ROP of NCA excludes the activated monomer mechanism, a dimerization reaction occurs to yield 2,5-diketopiperazine particularly at a low concentration of monomers. A type of NTA monomer was reported to polymerize under mild environments tolerant to moisture[16]. The synthetic approach of NTA monomers is similar to that of NCA, except N-ethoxythiocarbonyl amino acid is used as the precursor and phosphorous tribromide is used for cyclization reaction instead. Such tunable synthetic approaches greatly promote the development of stimuli-responsive polypeptoid-based materials for many applications.
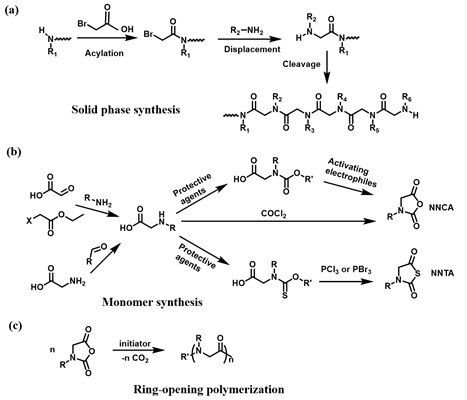
Scheme 1. Synthesis of polypeptoid. (a) “Submonomer” solid-phase synthesis, (b) Synthesis of N-substituted glycine N-carboxyanhydrides (NCA) and N-substituted N-thiocarboxyanhydrides (NTA) monomers, (c) NCA ring-opening polymerization.
2.2 Post-Polymerization Modification (PPM) Strategy
Considering the complex purification and protection–deprotection chemistry in the preparation of functional monomer strategy, post-polymerization modification of a well-defined polypeptoid precursor is more convenient. The key of the PPM route is to employ the effective coupling strategy for the pendant moieties to conjugate with the functional groups. “Click chemistry”, such as thiol-ene/yne addition chemistry and copper-catalyzed azide-alkyne click chemistry (CuAAC), has emerged as one of the most efficient strategies to obtain the stimuli-responsive polypeptoids in high yield. Schlaad et al. demonstrated the post modifications of poly(N-allyl glycine) (PNAG) and poly(N-propargyl glycine) (PNPgG) through thiol-ene chemistry to introduce various functional groups[17]. Sun and coworkers successfully prepared a family of thermoresponsive polypeptoids by combining ROP of NCA and thiol-ene/yne click chemistry[18][19][20]. Zhang et al. reported that the cyclic PNPgG homopolymer and copolymer can be modified with azides by CuAAC ( Figure 1)[11].
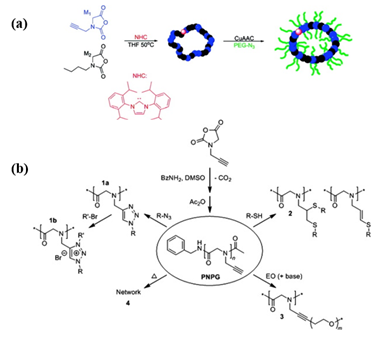
Figure 1. (a) The formation of cyclic brush-like polymers from the reaction of azido-terminated poly(ethylene glycol) (PEG) and cyclic poly(N-propargyl glycine)-ran-poly(N-butyl glycine) copolypeptoids bearing propargyl side chains. Reprinted with permission from[11]. Copyright 2011, American Chemical Society. (b) Synthesis of poly(N-propargyl glycine) and subsequent modifications of alkyne side chains. Reprinted with permission from[17]. Copyright 2015, Elsevier.
3. Thermoresponsive Polypeptoids
Due to the similarity to the chemical structures of a few typical thermoresponsive polymers such as polyacrylamides or poly(2-oxazoline)s, polypeptoids also show lower critical solution temperature (LCST) behavior in aqueous solution. In addition, the chemical structure of the side chains on the polypeptoid polymers can largely determine its thermoresponsive performances. Therefore, precise construction of the side-chain moieties is considered as an efficient strategy to prepare a variety of thermoresponsive polypeptoids. In this section, design and synthesis of thermoresponsive polypeptoids with different side chains will be discussed in detail.
3.1. Thermoresponsive Polypeptoids Containing Alkyl Side Chains
Polypeptoids bearing short aliphatic side chains such as methyl (C1), ethyl (C2), and 2-methoxyethyl N-substituents have good solubility while the polypeptoids with longer alkyl side chains (>C3) were found to be insoluble in water[21]. Schlaad et al. prepared a series of well-defined poly(N-alkyl glycine) with C3 N-substituents (i.e., n-propyl, allyl, propargyl, and isopropyl) by ring-opening polymerization of N-substituted glycine N-carboxyanhydrides (NCA) and then investigated the solubility of these polypeptoids[22]. They observed that except for the poly(N-propargyl glycine), the other three polypeptoids were soluble in water at lower temperature and became insoluble at higher temperature, which showed a typical LCST behavior. It was observed that the phase transition of these polypeptoids can be affected by the concentration, leading to various Tcp. Moreover, the broad transitions and pronounced hysteresis during the heating and cooling scans occurred in the solutions of poly(N-propyl glycine) (PNPG) and poly(N-isopropyl glycine) (PNiPG), while the PNAG solutions exhibited very sharp transitions that are similar to PNIPAM. In addition, they demonstrated that the molecular weight of polypeptoids show great influence on the cloud point temperatures (Tcp). The Tcp of PNAG decreases as the molecular weight increases, which may be attributed to a better hydration of shorter chains rather than the contributions of end groups. The opposite trend was observed for PNPG that can be explained by a poor shielding of short hydrophobic backbones in aqueous phase. The Tcp of the water-soluble polypeptoids was also dependent on the length of the side chains and polymer concentration (Figure 2). They also showed that the crystalline objects can be obtained from the solutions of PNPG and PNAG upon long-time annealing.Schlaad and coworkers also prepared thermoresponsive poly(N-propylglycine)-block-poly(N-methylglycine) (PNPG-b-PNMG) diblock copolypeptoid by successive ring-opening polymerization of NCA-initiated by primary amine[23]. Due to the presence of a thermoresponsive/ crystallizable PNPG block and a hydrophilic PNMG block, thermo-induced aggregation occurred in this case. At T > Tcp, the initial spherical aggregates morphology transformed into crystallization phase and then larger complex assemblies with flower-like, ellipsoidal, or irregular shapes were obtained.
Zhang and co-workers reported the reversible phase transition of cyclic and linear poly((N-ethyl glycine)-ran-(N-butyl glycine)) (c/l-P(NEG-r-NBG)) random copolymers in aqueous solution, both of which were synthesized by ROPs of the respective R-NCA monomers using either NHC or benzyl or butyl amine initiators[24]. It was observed that the cloud point temperature was shifted to higher temperature with increasing of N-ethyl glycine content with the Tcp ranging from 20 to 60℃. The topological architecture (cyclic and linear) has a great impact on the cloud point temperature. The Tcp of the cyclic copolypeptoids was lower than the linear analogues with the same composition because of the lower entropic loss. It was found that the cloud point temperature decreased with the increase of polymer concentration. The effect of the salt concentrations on the Tcp of the thermoresponsive polymers was further studied. The results revealed that the Tcp of copolypeptoids shifted to the lower temperature upon the addition of sodium salts, which was mostly consistent with the Hofmeister series (Figure 2).
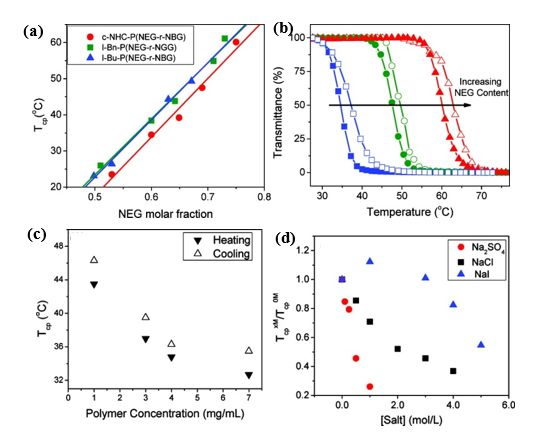
Figure 2. (a) Representative plots of transmittance at λ = 450 nm vs. temperature for the selected aqueous solutions of cyclic copolypeptoids c-NHC-P(NEG70-r-NBG47) (blue); c-NHC-P(NEG65-r-NBG30) (green); c-NHC-P(NEG101-r-NBG34) (red) (polymer concentration = 1.0 mg/mL; heating and cooling cycles are symbolized by the filled and unfilled symbols, respectively) (b) Plots of cloud point temperature (Tcp) versus the molar fraction of NEG segment in the cyclic and linear P(NEG-r-NBG) random copolymers bearing different end groups and their respective linearly fit curves, c-NHC-P(NEG70-r-NBG47) (blue); c-NHC-P(NEG65-r-NBG30) (green); c-NHC-P(NEG101-r-NBG34) (red). (c) Plot of Tcps of c-NHC-P(NEG55-r-NBG26) vs. the polymer concentration in water; (d) plot of Tcps of c-NHC-P(NEG62-r-NBG23) at various salt concentrations (TcpxM) relative to that with no salt (Tcp0M) vs. the salt concentration in water. Reprinted with permission from[24]. Copyright 2012, American Chemical Society.
Zhang et al. investigated crystallization-driven thermoreversible gelation of coil-crystalline cyclic and linear coil-crystalline diblock copolypeptoids, i.e., poly(N-methyl-glycine)-block-poly(N-decyl-glycine). The cyclic and linear coil-crystalline diblock copolypeptoids were observed to form free-standing gels consisting of entangled fibrils in methanol solutions (5–10 wt%) at room temperature. The gelation dissolved upon heating, which resulted in the transformation of fibrillar network morphology into an isotropic solution that was thermally reversible and mechanically nonreversible[25]. Further, they synthesized thermoresponsive ABC triblock copolypeptoids (i.e., poly(N-allyl glycine)-block-poly(N-methyl glycine)-block-poly(N-decylglycine)) that can exhibit sol-to-gel transition at elevated temperature in aqueous solutions at 2.5–10 wt% (Figure 3)[26]. In contrast to the reported ABC triblock copolymers, such as poly(ethylene-alt-propylene)-block-poly(ethylene oxide)-block-poly(N-isopropylacrylamide) (PON) and poly((propylenesulfide)-block-(N,N-dimethylacrylamide)-block-(N-isopropylacrylamide)) (PPS-b-PDMA-b-PNIPAAM) triblock copolymers, which are based on non-degradable polymers, the prepared ABC triblock copolypeptoids are biodegradable and cytocompatible. The gelation temperature and the mechanical properties of the hydrogels can be finely tuned by regulating the block copolymer composition and the polymer solution concentration.
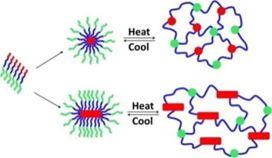
Figure 3. Schematic illustration of the gelation mechanism of the ABC triblock copolypeptoids poly(N-allyl glycine)-block-poly(N-methyl glycine)-block-poly(N-decyl glycine) in aqueous solution. Reprinted with permission from[26]. Copyright 2016, American Chemical Society.
Thermoresponsive bottlebrush copolypeptoids polynorbonene-graft-poly((N-ethyl glycine)-ran-(N-butyl glycine)) (PNor-g-P(NEG-r-NBG)) comprised of the liner P(NEG-r-NBG) random copolymer sidechains were also prepared by the Zhang group. The bottlebrushes were synthesized by the ring-opening metathesis polymerization (ROMP) of the norbornenyl-terminated P(NEG-r-NBG) macromonomers[27]. The cloud point transition of the bottlebrush copolymers in aqueous solution is similar to that of the linear macromonomers. However, compared to linear macromonomers, the Tcp of bottlebrush copolymers was slightly affected by the polymer architecture, but strongly dependent on the thermal history of the solution. On the other hand, the bottlebrush copolypeptoids exhibited the cloud point transition that was irrelevant to the solution thermal history with the addition of inorganic salt. They demonstrated that the cloud point transition was caused by the conformational restructuring of the bottlebrush copolypeptoids facilitated by the thermal annealing (Figure 4). In addition, the presence of salts favored hydrophobic collapse and intermolecular aggregation. Kuroda and Zhang et al. further explored the molecular mechanism of the thermoresponsive performance of polypeptoids by studying two polypeptoids (l-NHC-P(NEG-r-NBG) and c-NHC-P(NEG-r-NBG) with similar alkyl side chain compositions and different architecture[28]. The phase transition temperatures of cyclic and linear polypeptoids were 43 °C and 47 °C that were highly dependent on the polymer morphology of macromolecules with very similar solvent interaction but different conformational entropy. This suggested that the phase transition of these polypeptoids is dominated by the conformation of polymer backbone. They further demonstrated the molecular mechanism of the phase transition is associated with the variation of the polymer backbone conformation from a cis amide conformation to a random mixture of cis and trans amide conformations, which is significantly different from the mechanism of the hydration of the polymer resulted by a coil-to-globule transition.
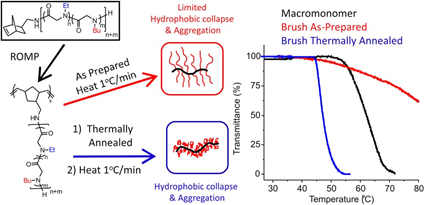
Figure 4. Structure of polypeptoid bottlebrush copolymers and schematic illustration of the level of aggregation in as-prepared or thermally annealed solutions of the polypeptoid bottlebrushes. Reprinted with permission from[27]. Copyright 2014, Royal Society of Chemistry.
Ling and coworkers synthesized a series of thermoresponsive random copolypeptoids poly(sarcosine-ran-N-butylglycine) (P(Sar-r-NBG)s) by the controlled copolymerization of sarcosine NTA (Sar-NTA) and N-butylglycine NTA (NBG-NTA) initiated by benzylamine[16][29]. They found that P(Sar-r-NBG)s show LCST with reversible phase transitions in aqueous solution. The solubility of these copolypeptoids was greatly affected by copolymer composition due to the hydrophobicity of PNBG. The Tcp can be tuned in a broad temperature range of 27–71 °C by controlling Sar molar fraction and degree of polymerization of random copolymers. It was observed that the Tcps of polypeptoids displayed a linear increase with the increase of Sar molar fraction, which indicates that the hydrophobic effect plays a crucial role in the thermoresponsive behavior of polypeptoids. Meanwhile, the Tcp depended slightly on polymer concentration and the MW, as well as salt additives. The Tcp increased with the decrease of MW, which is consistent with the other vinyl-based polymers[30] and different from the cyclic P(NEG-r-NBG)s[24]. Upon the addition of salt additives, the Tcp decreased, which coincided with the Hofmeister series. Such a result is comparable to the copolypeptoid P(NEG-r-NBG)s mentioned previously.
Recently, they synthesized both UV- and thermo-responsive diblock polypeptoids poly(sarcosine-ran-butylglycine)-block-PNB (P(Sar-r-NBG)-b-PNB) and triblock analogs via ROPs of a nitrobenzyl-containing NTA monomer (NB-NTA) with sarcosine NTA (Sar-NTA) and N-butylglycine NTA (NBG-NTA) comonomers[31]. The cleavage of nitrobenzyl ester side group occurred after the exposure at 254 nm UV irradiation, which results in the conversion of the lipophilic PNB block to hydrophilic polyiminodiacetic acid block. Simultaneously, the thermoresponsive P(Sar-r-NBG) block exhibits LCST phase transition behavior with temperature changing and their Tcp can be tuned by the composition of repeating units. Additionally, the block polypeptoids can self-assemble in water and form micelles with the PNB blocks as the core because of the amphiphilicity of both the diblock and triblock polypeptoids. Intriguingly, the morphology of spherical nanoparticles transformed into linear aggregates with the increase of concentration and thermal annealing of the samples (Figure 5).
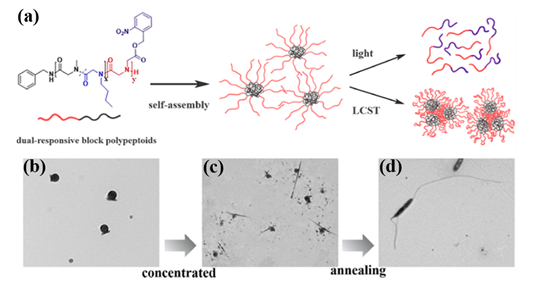
Figure 5. (a) Schematic illustration of dual-responsive block polypeptoids. (b) TEM micrograph of micelles formed by P(Sar0.51-r-NBG0.49)52-b-PNB7 in diluted aqueous solution (0.2 mg/mL). (c) TEM micrograph of structures formed by P(Sar0.51-r-NBG0.49)52-b-PNB7 in concentrated aqueous solution (5 mg/mL). (d) TEM micrograph of anisotropic structures formed by P(Sar0.51-r-NBG0.49)52-b-PNB7 in concentrated aqueous solution (5 mg/mL) after the annealing process. Reprinted with permission from[31]. Copyright 2020, American Chemical Society.
3.2. OEGlyated Thermoresponsive Polypeptoids
Poly(ethylene glycol) (PEG) is one of the most commonly used polymers in biotechnology which possesses many excellent properties such as non-toxicity, non-immunogenicity, hydrophilicity, high biocompatibility, and enhanced therapeutic efficacy. Due to the reversible hydration and dehydration of the oligo(ethylene glycol) (OEG) moieties upon temperature change, LCST-type thermoresponsive polymers can be obtained by introducing OEG pendants to the side chain[32]. Aoshima[33] and Lutz[34] investigated a range of biocompatible and thermoresponsive polymers based on (OEG) moieties. The Tcp of these copolymers could be tuned by adjusting the fraction of OEG units and molecular weight[35][36]. Many pegylated polypeptides have also been extensively investigated, several of them such as poly(L-glutamic acid) and poly(L-cysteine acid) show highly tunable thermal-responsive properties[37][38][39]. However, merely a few of pegylated polypeptides display thermoresponsive properties which may contribute to the fact that the solution properties of OEGylated polypeptides not only depended on substructure of side chains but also relied on their inherent secondary structures.
A few thermoresponsive polypeptoids have also been successfully prepared by the incorporation of OEG pendants on the side chain. Sun et al. synthesized a series of OEG-grafted poly (N-propargyl glycine) (PNPgGn-g-EGx) polypeptoids by a combination of ROP of N-propargylglycine N-carboxyanhydride initiated by primary amine and thiol-yne click chemistry[18]. They showed that the obtained polypeptoids displayed reversible thermoresponsive behavior in aqueous solution with a sharp phase transition, similar to PNIPAM. This differs distinctly from the previously reported pegylated polypeptoids synthesized by ROP of the corresponding N-carboxyanhydrides having oligomeric ethylene glycol side chains, which exhibits good water solubility[40][41][42]. They demonstrated that the introduction of both thioether moieties and OEG on the side chains may vary the amphiphilicity of the system that results in the thermoresponsive property. The influence of degree of polymerization (DP), the side-chain length, the polymer/salt concentration and the chain-ends on the Tcp was further observed in this work and it was found that the Tcp of the pegylated polypeptoids could be highly tuned by adjusting these factors (Figure 6). The Tcp of the polypeptoids increases with the increase of DP, which may be attributed to the short polymer chains that enables increased exposure of hydrophobic backbones to the water phase. In contrast, the polypeptides with similar OEG side-chains exhibited the opposite tendency. This is possibly because with the DP of the polymer increasing, the interaction between polymer chain and water phase reduced due to the increase of the content of a-helix, causing the lower Tcp[43]. Therefore, it is conceivable that the secondary conformation plays an important role on the thermoresponsive properties of polymers. They utilized different initiators to mediate the polymerization of the monomers and obtained PNPgGn-g-EGx with different chain ends, polypeptoid with HEXNH2 as initator is denoted as HEX-PNPgGn-g-EGx. In all cases, the Tcps of polypeptoids are comparable, which indicates that the chain-ends have negligible effect on Tcp[44]. It was also observed that the HEX-PNPgGn-g-EG3 displays a higher Tcp than that of HEX-PNPgGn-g-EG2 at same DP, which is because of the better hydration ability of the polymer with longer OEG units, irrespective of the backbone structure. Various pegylated polypeptoids can be obtained by adjusting the molar ratios of a mixture of thiol terminated OEG molecules to change the chemical compositions of the pendant side chains. They further synthesized a kind of pegylated polypeptoid that exhibited both reversible thermoresponsive and redox-responsive behaviors in aqueous solution[19]. It was found that the oxidation/reduction of pendant thioethers on the side chain had a great influence on the Tcp which providing a second stimulus to tune phase transition behavior.
Lately, they reported a stepwise crystallization-driven self-assembly (CDSA) process thermally initiated from amphiphilic poly(N-allyl glycine)-block-poly(N-octyl glycine) (PNAG-b-PNOG) diblock copolypeptoid conjugated with thiol-terminated triethylene glycol monomethyl ethers ((PNAG-g-EG3)-b-PNOG) in aqueous solution. The diblock copolypeptoids show a reversible LCST behavior by incorporating of thermosensitive PNAG-g-EG3 segments, by which the thermostimulus triggers the collapse of hydrophilic chains to promote the orderly fabrication of the crystalline PNOG block upon heating[45]. Due to the presence of a confined domain caused by crystalline PNOG, the morphology transition of the assemblies is irreversible upon a heating-cooling cycle, in which different nanostructured assemblies can be obtained by the same polymer.
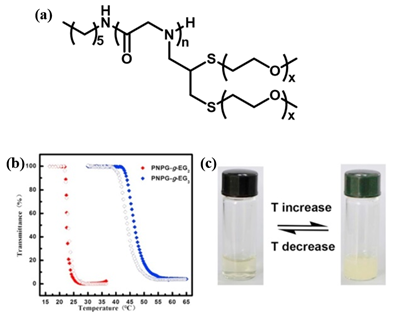
Figure 6. (a) Structure of HEX-PNPgGn-g-EGx. (b) Plots of transmittance as a function of temperature for aqueous solutions (2 mg/mL) of HEX-PNPgGn-g-EGx. Filled symbol: heating ramp, open symbol: cooling ramp. (c) The photographs of HEX-PNPgGn-g-EGx aqueous solution showing reversible phase transition. Reprinted with permission from[18]. Copyright 2018, Elsevier.
3.3. Charge-Determined Thermoresponsive Polypeptoids
The thermoresponsive polymers possessing both LCST and UCST show desired dual transitions in a specific range of environmental conditions, which offer great potential for smart materials. However, the polymers that can exhibit tunable phase transitions with both LCST and UCST behaviors in aqueous solution have rarely been reported. This is because delicate control over the balance of a variety of factors is typically required. Wu et al. prepared well-defined thermoresponsive copolymers by copolymerization of N-acryloylglycinamide and diacetone acrylamine, which have either LCST or UCSTtype transitions, depending on the compositions, degree of polymerization (DP), polymer concentration, and so on[46]. Wang et al. reported a type of architecture-determined thermoresponsive copolymer with a wide range of controllable LCST and UCST behavior by changing the chain length of poly(vinyl alcohol) grafted on poly(p-dioxanone)[47]. However, such investigations on polypeptoids with dual thermoresponsivity had not been reported until Sun et al. prepared a series of charge-determined ionic polypeptoids with tunable thermoresponsive property by a combination of ring-opening polymerization and click chemistry, which was unlike the previous pegylated nonionic polypeptoids that exhibits LCST behavior[20]. The polypeptoids exhibited either LCST or UCST type behavior, depending on the charge state of the side chains of polypeptoids (Figure 7). Meanwhile, the phase transition of the polypeptoids was also sensitive to PH in aqueous solution which was attributed to the presence of charge. The side-chain architecture, the degree of polymerization (DP), and the polymer concentration were found to have significant effect on the solution properties of the polypeptoids.
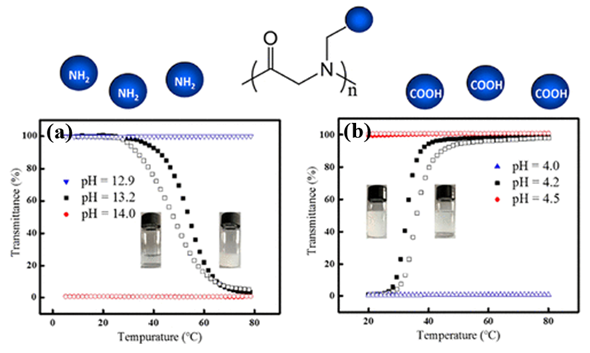
Figure 7. Plots of transmittance as a function of temperature for aqueous solutions (a) PNPG41-(COOH)2 (2 mg/mL) at different pH. Filled symbol: cooling ramp; open symbol: heating ramp. (b) PNAG79-NH2 (10 mg/mL) at different pH. Filled symbol: heating ramp; open symbol: cooling ramp. Reprinted with permission from[20]. Copyright 2018, American Chemical Society.
In recent work, by introducing alkyl or ethylene glycol (EG) units, a systematic study on thermo-responsive polypeptoid exhibited both LCST and UCST-type behavior was prepared into ionic polypeptoids via combining ROP and thiol-ene/yne click chemistry[48]. The phase transition temperature that is dependent on the chain length of pendant groups, the chemical composition, and the species of ionic moieties can be tuned in the range of 29–55 °C. Intriguingly, they demonstrate that the UCST behavior of polypeptoids can be regulated by the hydrophilic EG group, while the LCST behavior can be tuned by the hydrophobic residues.
4. Applications
Various applications of thermoresponsive polymers have been studied in many research fields such as controlled drug delivery, nanoreactors, smart coatings, and sensors[49][50][51]. PNIPAM as the most widely studied thermoresponsive polymer has been extensively used in many biomedical applications such as thermoresponsive cell culture dish modified with PNIPAM on which the adhesion and detachment of the cell can be controlled by changing the temperature. The intact cell monolayer can be successfully obtained on the thermoresponsive cell culture dish[52]. Lu et al. synthesized a series of thermoresponsive polypeptides incorporated with various functional side groups[53]. They found that the polypeptides showed excellent biodegradability, biocompatibility, and mechanical properties in in vitro adhesion tests with no cytotoxicity. The polypeptides also showed perfect hemostatic properties and healing effects which are expected to be potential candidates for medical applications, such as tissue adhesives, tissue engineering, and arresting bleeding. Thermoresponsive polypeptoids with temperature-controlled solubility in aqueous solution are promising candidates for biomedical applications due to their good biocompatibility and degradability. Ling et al. investigated the potential of both UV- and thermo-responsive diblock polypeptoids used as a drug delivery system and found that both nile red and pyrene as model hydrophobic guest molecules were encapsulated and released by UV irradiation triggered or a local change in environment [62]. Zhang and coworkers demonstrated that water-soluble enzymes can be easily encapsulated in the ABC triblock copolypeptoid hydrogels for extended period of time with the retained enzymatic activity. The hydrogels show low cytotoxicity towards human adipose-derived stem cells (hASCs) and exhibit bioactivity in modulating the chondrogenesis biomarker gene expression of hASCs, which indicate the potential utilization of the polypeptoid hydrogel as tissue engineering material [57]. The monodisperse, superparamagnetic iron oxide nanoparticles (SPION) modified with a thermoresponsive polypeptoid poly(N-methylglyine)-ran-poly(N-butylglycine) (PNMG-r-PNBG) brush was prepared via controlled surface-initiated polymerization of N-substituted NCAs[54]. The prepared SPION exhibiting excellent biocompatibility, stability, and high functionality can be used in many applications such as magnetic molecular extraction and magnetic resonance imaging. Garno et al. utilized thiol-ene click reaction to conjugate thermoresponsive copolypeptoid poly((N-ethylglycine)32-ran-(N-butylglycine)17) P(NEG32-r-NBG17) to 10-undecenyltrichlorosilane (UTS) nanopatterns to prepare nanostructures of copolypeptoid brushes on a silicon substrate[55]. The reversible phase transitions of nanopatterns of copolypeptoids were investigated in situ with atomic force microscopy (AFM) and it was found that the nanostructures shrink in size to form collapsed patterns after heating. On the contrary, the polymer strands stretch out to form taller patterns upon cooling. They demonstrated that the hydrophobic characteristics of surface coatings can be greatly affected by the phase transitions of thermoresponsive polymers anchored to surfaces, which in turn can be used to regulate the adsorption or rejection tendency of the coating to biomacromolecules. Smart coatings of P(NEG32-r-NBG17) with temperature-dependent surface changes will enable the design of surface-based sensors.
References
- Gianine M. Figliozzi; Richard Goldsmith; Simon C. Ng; Steven C. Banville; Ronald N. Zuckermann; [25] Synthesis of N-substituted glycine peptoid libraries. Methods in Enzymology 1996, 267, 437-447, 10.1016/s0076-6879(96)67027-x.
- Niklas Gangloff; Juliane Ulbricht; Thomas Lorson; Helmut Schlaad; Robert Luxenhofer; Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering. Chemical Reviews 2015, 116, 1753-1802, 10.1021/acs.chemrev.5b00201.
- Donghui Zhang; Samuel H. Lahasky; Li Guo; Chang-Uk Lee; Monika LaVan; Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833-5841, 10.1021/ma202319g.
- Alexander Birke; David Huesmann; Annette Kelsch; Martin Weilbächer; Jing Xie; Matthias Bros; Tobias Bopp; Christian Becker; Katharina Landfester; Matthias Barz; et al. Polypeptoid-block-polypeptide Copolymers: Synthesis, Characterization, and Application of Amphiphilic Block Copolypept(o)ides in Drug Formulations and Miniemulsion Techniques. Biomacromolecules 2014, 15, 548-557, 10.1021/bm401542z.
- Timothy J. Deming; Synthesis of Side-Chain Modified Polypeptides. Chemical Reviews 2015, 116, 786-808, 10.1021/acs.chemrev.5b00292.
- R. J. Simon; R. S. Kania; R. N. Zuckermann; V. D. Huebner; D. A. Jewell; S. Banville; S. Ng; L. Wang; S. Rosenberg; C. K. Marlowe; et al. Peptoids: a modular approach to drug discovery.. Proceedings of the National Academy of Sciences 1992, 89, 9367-9371, 10.1073/pnas.89.20.9367.
- Ronald N. Zuckermann; Peptoid origins. Biopolymers 2010, 96, 545-555, 10.1002/bip.21573.
- Li Guo; Donghui Zhang; Cyclic Poly(α-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-SubstitutedN-Carboxylanhydrides. Journal of the American Chemical Society 2009, 131, 18072-18074, 10.1021/ja907380d.
- Xinfeng Tao; Chao Deng; Jun Ling; PEG-Amine-Initiated Polymerization of SarcosineN-Thiocarboxyanhydrides Toward Novel Double-Hydrophilic PEG-b-Polysarcosine Diblock Copolymers. Macromolecular Rapid Communications 2014, 35, 875-881, 10.1002/marc.201400066.
- Li Guo; Samuel H. Lahasky; Kushal Ghale; Donghui Zhang; N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydrides toward Poly(α-peptoid)s: Kinetic, Mechanism, and Architectural Control. Journal of the American Chemical Society 2012, 134, 9163-9171, 10.1021/ja210842b.
- Samuel H. Lahasky; Wilson K. Serem; Li Guo; Jayne C. Garno; Donghui Zhang; Synthesis and Characterization of Cyclic Brush-Like Polymers byN-Heterocyclic Carbene-Mediated Zwitterionic Polymerization ofN-PropargylN-Carboxyanhydride and the Grafting-to Approach. Macromolecules 2011, 44, 9063-9074, 10.1021/ma201948u.
- Xinfeng Tao; Botuo Zheng; Tianwen Bai; Baoku Zhu; Jun Ling; Hydroxyl Group Tolerated Polymerization of N-Substituted Glycine N-Thiocarboxyanhydride Mediated by Aminoalcohols: A Simple Way to α-Hydroxyl-ω-aminotelechelic Polypeptoids. Macromolecules 2017, 50, 3066-3077, 10.1021/acs.macromol.7b00309.
- Xinfeng Tao; Botuo Zheng; Tianwen Bai; Min-Hui Li; Jun Ling; Polymerization of N-Substituted Glycine N-Thiocarboxyanhydride through Regioselective Initiation of Cysteamine: A Direct Way toward Thiol-Capped Polypeptoids. Macromolecules 2018, 51, 4494-4501, 10.1021/acs.macromol.8b00259.
- Joshua W. Robinson; Helmut Schlaad; A versatile polypeptoid platform based on N-allyl glycine. Chemical Communications 2012, 48, 7835-7837, 10.1039/c2cc33881e.
- Brandon A. Chan; Sunting Xuan; Ang Li; Jessica M. Simpson; Garrett L. Sternhagen; Tianyi Yu; Omead A. Darvish; Naisheng Jiang; Donghui Zhang; Polypeptoid polymers: Synthesis, characterization, and properties. Biopolymers 2017, 109, e23070, 10.1002/bip.23070.
- Xinfeng Tao; Yangwei Deng; Zhiquan Shen; Jun Ling; Controlled Polymerization of N-Substituted Glycine N-Thiocarboxyanhydrides Initiated by Rare Earth Borohydrides toward Hydrophilic and Hydrophobic Polypeptoids. Macromolecules 2014, 47, 6173-6180, 10.1021/ma501131t.
- Christian Secker; Joshua W. Robinson; Helmut Schlaad; Alkyne-X modification of polypeptoids. European Polymer Journal 2015, 62, 394-399, 10.1016/j.eurpolymj.2014.08.028.
- Jiliang Tian; Jing Sun; Zhibo Li; Biomimetic pegylated polypeptoids with thermoresponsive properties. Polymer 2018, 138, 132-138, 10.1016/j.polymer.2018.01.034.
- Xiaohui Fu; Jiliang Tian; Zheng Li; Jing Sun; Zhibo Li; Dual‐responsive pegylated polypeptoids with tunable cloud point temperatures. Biopolymers 2018, 110, e23243, 10.1002/bip.23243.
- Chao Xing; Zhekun Shi; Jiliang Tian; Jing Sun; Zhibo Li; Charge-Determined LCST/UCST Behavior in Ionic Polypeptoids. Biomacromolecules 2018, 19, 2109-2116, 10.1021/acs.biomac.8b00240.
- Corinna Fetsch; Arlett Grossmann; Lisa Holz; Jonas F. Nawroth; Robert Luxenhofer; Polypeptoids fromN-Substituted GlycineN-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution. Macromolecules 2011, 44, 6746-6758, 10.1021/ma201015y.
- Joshua W. Robinson; Christian Secker; Steffen Michael Weidner; Helmut Schlaad; Thermoresponsive Poly(N-C3 glycine)s. Macromolecules 2013, 46, 580-587, 10.1021/ma302412v.
- Christian Secker; Antje Völkel; Brigitte Tiersch; Joachim Koetz; Helmut Schlaad; Thermo-Induced Aggregation and Crystallization of Block Copolypeptoids in Water. Macromolecules 2016, 49, 979-985, 10.1021/acs.macromol.5b02481.
- Samuel H. Lahasky; Xiaoke Hu; Donghui Zhang; Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture. ACS Macro Letters 2012, 1, 580-584, 10.1021/mz300017y.
- Chang-Uk Lee; Lu Lu; Jihua Chen; Jayne C. Garno; Donghui Zhang; Crystallization-Driven Thermoreversible Gelation of Coil-Crystalline Cyclic and Linear Diblock Copolypeptoids. ACS Macro Letters 2013, 2, 436-440, 10.1021/mz300667n.
- Sunting Xuan; Chang-Uk Lee; Cong Chen; Andrew B. Doyle; Yueheng Zhang; Li Guo; Vijay T. John; Daniel J Hayes; Donghui Zhang; Thermoreversible and Injectable ABC Polypeptoid Hydrogels: Controlling the Hydrogel Properties through Molecular Design. Chemistry of Materials 2016, 28, 727-737, 10.1021/acs.chemmater.5b03528.
- Samuel H. Lahasky; Lu Lu; Wayne A. Huberty; Jinbao Cao; Li Guo; Jayne C. Garno; Donghui Zhang; Synthesis and characterization of thermo-responsive polypeptoid bottlebrushes. Polymer Chemistry 2013, 5, 1418-1426, 10.1039/c3py01356a.
- Jianbo Ma; Sunting Xuan; Abby C. Guerin; Tianyi Yu; Nghui Zhang; Daniel G. Kuroda; Unusual molecular mechanism behind the thermal response of polypeptoids in aqueous solutions. Physical Chemistry Chemical Physics 2017, 19, 10878-10888, 10.1039/c6cp08536a.
- Xinfeng Tao; Jianwei Du; Youxiang Wang; Jun Ling; Polypeptoids with tunable cloud point temperatures synthesized from N-substituted glycine N-thiocarboxyanhydrides. Polymer Chemistry 2015, 6, 3164-3174, 10.1039/c5py00191a.
- Stephan Salzinger; Uwe B. Seemann; Andriy Plikhta; Bernhard Rieger; Poly(vinylphosphonate)s Synthesized by Trivalent Cyclopentadienyl Lanthanide-Induced Group Transfer Polymerization. Macromolecules 2011, 44, 5920-5927, 10.1021/ma200752d.
- Yao Li; Jessica C. Tom; Philip Biehl; Jun Ling; Felix H. Schacher; Block Polypeptoids: Synthesis, Characterization, and Response Toward Irradiation with UV Light and Temperature. Macromolecules 2020, 53, 5218-5226, 10.1021/acs.macromol.0c00654.
- Guillaume G. Hedir; Maria C. Arno; Marvin Langlais; Jonathan T. Husband; Rachel K. O'reilly; Andrew P. Dove; Poly(oligo(ethylene glycol) vinyl acetate)s: A Versatile Class of Thermoresponsive and Biocompatible Polymers. Angewandte Chemie International Edition 2017, 56, 9178-9182, 10.1002/anie.201703763.
- Daisuke Aoki; Hiroharu Ajiro; Thermoresponsive Polyurethane Bearing Oligo(Ethylene Glycol) as Side Chain Without Polyol at Polymer Backbone Achieved Excellent Hydrophilic and Hydrophobic Switching. Macromolecular Rapid Communications 2018, 39, e1800239, 10.1002/marc.201800239.
- Jean-François Lutz; Ann Hoth; Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules 2006, 39, 893-896, 10.1021/ma0517042.
- Yanzhi Xu; Mengxiang Zhu; Minjie Li; Ying Ling; Haoyu Tang; Synthesis and LCST-type phase behavior of water-soluble polypeptide with Y-shaped and charged side-chains. Polymer Chemistry 2016, 7, 1922-1930, 10.1039/C5PY01991E.
- Yilong Cheng; Chaoliang He; Chunsheng Xiao; Jianxun Ding; Xiuli Zhuang; Xuesi Chen; Versatile synthesis of temperature-sensitive polypeptides by click grafting of oligo(ethylene glycol). Polymer Chemistry 2011, 2, 2627-2634, 10.1039/c1py00281c.
- Chongyi Chen; Zhaohui Wang; Zhibo Li; Thermoresponsive Polypeptides from Pegylated Poly-l-glutamates. Biomacromolecules 2011, 12, 2859-2863, 10.1021/bm200849m.
- Xiaohui Fu; Yong Shen; Wenxin Fu; Zhibo Li; Thermoresponsive Oligo(ethylene glycol) Functionalized Poly-l-cysteine. Macromolecules 2013, 46, 3753-3760, 10.1021/ma400678w.
- Jessica R. Kramer; Timothy J. Deming; Multimodal Switching of Conformation and Solubility in Homocysteine Derived Polypeptides. Journal of the American Chemical Society 2014, 136, 5547-5550, 10.1021/ja500372u.
- Sunting Xuan; Sudipta Gupta; Xin Li; Markus Bleuel; Gerald J. Schneider; Donghui Zhang; Synthesis and Characterization of Well-Defined PEGylated Polypeptoids as Protein-Resistant Polymers. Biomacromolecules 2017, 18, 951-964, 10.1021/acs.biomac.6b01824.
- Jing Sun; Gregory M. Stone; Nitash P. Balsara; Ronald N. Zuckermann; Structure–Conductivity Relationship for Peptoid-Based PEO–Mimetic Polymer Electrolytes. Macromolecules 2012, 45, 5151-5156, 10.1021/ma300775b.
- Andrea R. Statz; Robert J. Meagher; Annelise E. Barron; Zhuojun Huang; New Peptidomimetic Polymers for Antifouling Surfaces. Journal of the American Chemical Society 2005, 127, 7972-7973, 10.1021/ja0522534.
- Shusheng Zhang; Chongyi Chen; Zhibo Li; Effects of molecular weight on thermal responsive property of pegylated poly-l-glutamates. Chinese Journal of Polymer Science 2013, 31, 201-210, 10.1007/s10118-013-1218-7.
- Yan Xia; Xiangchun Yin; Nicholas A. D. Burke; Harald D. H. Stöver; Thermal Response of Narrow-Disperse Poly(N-isopropylacrylamide) Prepared by Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5937-5943, 10.1021/ma050261z.
- Zhiwei Wang; Min Lin; Colin Bonduelle; Rongye Li; Zhekun Shi; Chenhui Zhu; Sébastien Lecommandoux; Zhibo Li; Jing Sun; Thermoinduced Crystallization-Driven Self-Assembly of Bioinspired Block Copolymers in Aqueous Solution. Biomacromolecules 2020, 21, 3411-3419, 10.1021/acs.biomac.0c00844.
- Wenhui Sun; Zesheng An; Peiyi Wu; UCST or LCST? Composition-Dependent Thermoresponsive Behavior of Poly(N-acryloylglycinamide-co-diacetone acrylamide). Macromolecules 2017, 50, 2175-2182, 10.1021/acs.macromol.7b00020.
- Gang Wu; Si-Chong Chen; Qi Zhan; Yu-Zhong Wang; Well-Defined Amphiphilic Biodegradable Comb-Like Graft Copolymers: Their Unique Architecture-Determined LCST and UCST Thermoresponsivity. Macromolecules 2011, 44, 999-1008, 10.1021/ma102588k.
- Xiaohui Fu; Chao Xing; Jing Sun; Tunable LCST/UCST-Type Polypeptoids and Their Structure–Property Relationship. Biomacromolecules 2020, 21, 4980-4988, 10.1021/acs.biomac.0c01177.
- Jinming Hu; Shiyong Liu; Responsive Polymers for Detection and Sensing Applications: Current Status and Future Developments. Macromolecules 2010, 43, 8315-8330, 10.1021/ma1005815.
- Lianxiao Liu; Wen Li; Kun Liu; Jiatao Yan; Guixia Hu; Afang Zhang; Comblike Thermoresponsive Polymers with Sharp Transitions: Synthesis, Characterization, and Their Use as Sensitive Colorimetric Sensors. Macromolecules 2011, 44, 8614-8621, 10.1021/ma201874c.
- King Hang Aaron Lau; Peptoids for biomaterials science. Biomaterials Science 2014, 2, 627-633, 10.1039/c3bm60269a.
- Noriko Yamada; Teruo Okano; Hideaki Sakai; Fumiko Karikusa; Yoshio Sawasaki; Yasuhisa Sakurai; Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Die Makromolekulare Chemie, Rapid Communications 1990, 11, 571-576, 10.1002/marc.1990.030111109.
- Dedai Lu; Hongsen Wang; Ting’E Li; Yunfei Li; Xiangya Wang; Pengfei Niu; Hongyun Guo; Shaobo Sun; Xiaoqi Wang; Xiaolin Guan; et al.Hengchang MaZiqiang Lei Versatile Surgical Adhesive and Hemostatic Materials: Synthesis, Properties, and Application of Thermoresponsive Polypeptides. Chemistry of Materials 2017, 29, 5493-5503, 10.1021/acs.chemmater.7b00255.
- Steffen Kurzhals; Barbara Pretzner; Erik Reimhult; Ronald Zirbs; Thermoresponsive Polypeptoid-Coated Superparamagnetic Iron Oxide Nanoparticles by Surface-Initiated Polymerization. Macromolecular Chemistry and Physics 2017, 218, 1700116, 10.1002/macp.201700116.
- Lu Lu; Samuel H. Lahasky; Gregory T. McCandless; Donghui Zhang; Jayne C. Garno; Thermoresponsive Behavior of Polypeptoid Nanostructures Investigated with Heated Atomic Force Microscopy: Implications toward the Development of Smart Coatings for Surface-Based Sensors. ACS Applied Nano Materials 2019, 2, 7617-7625, 10.1021/acsanm.9b01715.

