 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shama Mirza | + 3808 word(s) | 3808 | 2020-12-18 08:53:03 | | | |
| 2 | Bruce Ren | Meta information modification | 3808 | 2020-12-28 03:14:21 | | |
Video Upload Options
Glioblastoma is one of the most common and detrimental forms of solid brain tumor, with over 10,000 new cases reported every year in the United States. Despite aggressive multimodal treatment approaches, the overall survival period is reported to be less than 15 months after diagnosis. A widely used approach for the treatment of glioblastoma is surgical removal of the tumor, followed by radiotherapy and chemotherapy. While there are several drugs available that are approved by the Food and Drug Administration (FDA), significant efforts have been made in recent years to develop new chemotherapeutic agents for the treatment of glioblastoma.
1. Introduction
Gliomas refer to all forms of intra-axial tumors that originate from glial cells of the central nervous system (CNS). They are the most common type of CNS tumors, representing about 80% of all malignant brain tumors [1][2]. Historically, they include types of cells that share similar histological characteristics, such as astrocytomas (high-grade astrocytomas are denominated glioblastomas), brain stem gliomas, ependymomas, oligodendrogliomas, optic pathway gliomas, and mixed gliomas [3][4]. This method of categorization helps to understand the histological features of gliomas; however, it does not provide information on the malignancy of a tumor. Meanwhile, rapid exploration in the past decade has provided significant insight not only for understanding the mechanisms of the neoplasm on a molecular basis, but also in designing new anticancer treatments. Therefore, in 2014, the International Society of Neuropathology included molecular information on top of the histological characteristics in brain tumor diagnoses [5][6][7]. This led to substantial modifications to the World Health Organization Classification of Tumors of the CNS (CNS WHO) in 2016 [5][6]. The updated CNS WHO further classified gliomas into grades (Grade I, II, III, and IV) based on pathological evaluation using molecular information on the malignancy level of the neoplasm. This subcategorization is particularly influential in clinical settings, as it can assist in determining the type of treatment(s) for patients. Grade I tumors are neoplasms with low proliferation rates that can be cured by surgery alone. On the other hand, grade II tumors are invasive and often recur despite low proliferative potential. Grade III tumors are generally malignant tumors with histological confirmation that exhibit anaplasia and rapid mitotic cell division, while grade IV gliomas are of the most advanced grade and are malignant tumors that have the poorest prognosis, with high potential for fatal outcome [5][6][8].
The most common and yet most deleterious grade IV glioma subtype is glioblastoma [9]. According to the Central Brain Tumor Registry of the United States (CBTRUS) Statistical Report 2011–2015, glioblastomas constitute about 57% of the average annual age-adjusted incidence rate of all neuroepithelial tumors and about 48% of all malignant brain and CNS tumors. It has been noted that the incidence rate of glioblastoma tumors is 1.58 times higher in the male population compared to females in the United States [1]. Despite aggressive multimodal treatment, due to the detrimental nature and quick progression (median survival of about 15 months) of glioblastomas, it is almost impossible to cure these patients [2]. Moreover, the heterogeneous nature of glioblastomas makes it extremely challenging to develop an effective therapeutic approach with a uniform outcome for all patients [2][10].
Current standard glioblastoma treatment is multimodal in nature, involving surgery, radiotherapy, and chemotherapy. Surgery for glioblastoma aims for a maximal and safe resection of the tumor. Maximal resection not only helps to relieve the mass pressure in the brain but also prolong overall survival (OS) rate, as shown in a recent study by Yamaguchi et al. [11]. They reported that maximal resection for glioblastoma increases OS compared to incomplete resection [11][12][13][14]. Furthermore, the technological advancement of surgical therapy aided by fluorescence visualization with 5-aminolevulinic acid, the navigation-guided fence post procedure, and intraoperative MRI has facilitated maximal and almost complete resection of tumors [12][13][14]. After surgery, most patients undergo radiotherapy and chemotherapy concurrently. The current standard radiotherapy dosage regimen is 2 Gy per fraction per day for 5 days a week, continuously for 6 weeks, with a total dosage of 60 Gy [2]. Early radiotherapy soon after surgery has shown to increase progression-free survival (PFS). However, for OS no significant improvement has been shown [14]. Surgical and radiotherapeutic management of the disease has been extensively reviewed elsewhere [15][16][17][18][19][20][21][22].
2. Pathogenesis
Understanding the pathogenesis plays a key role not only in identifying disease biomarkers but also in designing and developing potential chemotherapeutic agents. Herein, we discuss the nine most promising signaling pathways that are involved in pathogenesis, and the possibility of targeting specific components of these pathways for the development of chemotherapeutic agents for glioblastoma.
2.1. IDH Mutation
Isocitrate dehydrogenase (IDH) is an enzyme that plays a central role in the citric acid cycle. IDH has three isoforms: IDH1, IDH2, and IDH3. IDH1 is found in peroxisomes and the cytoplasm, while IDH2 and IDH3 are found in the mitochondrial matrix. Through oxidative decarboxylation by IDH1 and IDH2, isocitrate and NADP+ are converted to α-ketoglutarate (α-KG), NADPH, and carbon dioxide [23]. This takes place in a reversible, multistep process that starts with the oxidation of isocitrate to form oxalosuccinate, which is then decarboxylated to form α-KG, a cofactor for several enzymes (Figure 1) [24].
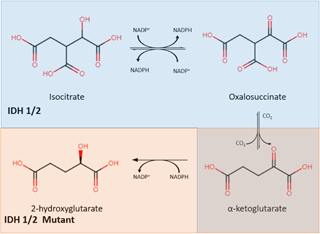
Figure 1. The IDH1/2 enzyme converts isocitrate into α-ketoglutarate (shown in the blue) while the mutant IDH1/2 enzyme converts α-ketoglutarate into 2-hydroxyglutarate (shown in orange). IDH: isocitrate dehydrogenase.
Mutations in IDH were found to be in almost all cases of secondary glioblastoma, as reported by Parsons et al. IDH mutations exist in high numbers in secondary glioblastomas and grade II and III gliomas but are rare in primary glioblastomas [25]. The IDH mutation involves both a loss and gain of regular enzymatic function [26]. It leads to a decrease in its binding affinity for isocitrate, preventing the conversion of isocitrate to α-KG. In addition, IDH mutation also increases its binding affinity for NADPH, which results in incomplete reaction by only reducing α-KG without carboxylation, forming 2-hydroxyglutarate (2-HG) instead of α-KG. The abnormal accumulation of 2-HG, an oncometabolite, is responsible for cancerogenesis [27]. This discovery resulted in mutant IDH (mIDH) inhibitors being identified as a new group of targeted cancer therapies which help to separate proliferating cancer cells. Popovici-Muller et al. reported that the mIDH1 inhibitor AGI-5198 was successful in 2-HG inhibition, and hindered the growth of mIDH1 glioma cells in vivo [28]. Optimization of AGI-5198 led to the finding of AG-120, which became the first mIDH1 inhibitor to achieve clinical proof-of-concept in human trials [28]. A selective R132H-IDH1 inhibitor, AG-5198, was discovered to almost completely block the ability of mIDH1 to produce 2-HG, and induced expression of genes involved in gliogenesis [29].
Results from clinical studies show that AG-221 (a selective inhibitor of mIDH2) has a promising inhibitory effect against advanced solid tumors [30]. There is an ongoing phase I clinical trial (NCT03343197) with AG-120 (mIDH1 inhibitor) and AG881 (non-specific IDH inhibitor). The objective of this trial is to understand the role of AG-120 and AG881 in the suppression of 2-HG by comparing the concentration of 2-HG in resected and treated tumors from IDH1 mutant glioma patients with the concentration of 2-HG in untreated tumor. Currently, two other chemotherapeutic agents, FT-2102 (a selective mIDH1 inhibitor) and IDH305 (an IDH1(R132H) inhibitor), are also in clinical trials (NCT03684811, NCT02381886). The objective of these clinical trials is to determine the dose-limiting toxicities (DLTs).
2.2. Notch Pathway
The Notch signaling plays an important role in cell differentiation, proliferation, and apoptotic events in different cell types and tissues, including neurons of the CNS. It is necessary to ensure that neural stem cells are promoted towards becoming glial cells instead of differentiating into another form [31]. Due to its key role in cell processes, it is easy for Notch signaling to deviate towards tumorigenesis.
There are four receptors involved in this pathway; Notch-1, Notch-2, Notch-3, and Notch-4. Notch-1 is found to be either a tumor suppressor or an oncogene based on the tissue type. Moreover, it has been found to be associated with glioma progression to determine the malignant phenotype of glioma. Notch-2, on the other hand, was identified as a prognostic marker for glioma along with Notch-3, which also promotes glioma cell proliferation. Lastly, Notch-4 was found to correlate with tumor aggressiveness [32].
Studies have shown the Notch pathway to be a potential and effective target in stem-like glioma cells, which were found to express Notch family genes [33]. In general, drugs inhibiting the Notch pathway are classified into three categories: α-secretase inhibitors, γ-secretase inhibitors, and other molecules. A detailed discussion of different classes of inhibitors and their biological effects has been published by Bazzoni et al. [34]. Ying et al. studied glioblastoma stem-like cell response to all-trans retinoic acid (RA) treatment. They found that RA can downregulate neurosphere cell expression of the Notch pathway targets Hes2, Hey1, and Hey2. When treated with RA, Notch receptor intracellular domain (NICD1) is forced to rescue glioblastoma neurospheres, thus causing inhibition of Hes2, Hey1, and Hey2. They concluded that this is an indication of RA affecting glioblastoma stem-like cells towards cell growth arrest, differentiation, and stem cell pool loss [35].
Similarly, Hovinga et al. performed a study on the relationship of neurosphere formation and CD133+ cells. It has been shown in the past that CD133+ cells are capable of self-renewal via the Notch pathway. Consequently, they discovered that Notch inhibition led to a decrease of neurosphere formation and CD133+ cells in glioblastoma while promoting an increased sensitivity to radiation [36]. Fan et al. studied glioblastoma neurosphere formation and Notch-2, which increases tumor cell growth. They demonstrated that inhibition of the Notch pathway, using gamma-secretase inhibitors, reduced glioblastoma neurosphere engraftment in vivo, which caused mice to live longer [33]. These studies indicate that inhibition of the Notch pathway is a potential therapeutic strategy to treat glioblastoma [33][36]. Currently there is one Notch inhibiting agent, CB-103, in a phase I/IIA clinical trial (NCT03422679) against metastatic solid tumors. The current primary outcome measures of the trial are to determine DLTs and antitumor efficacy.
2.3. Ceramide Signaling
Acid ceramidase (ASAH1) is an enzyme that metabolizes ceramides into sphingosine and free fatty acids (Figure 2). Ceramides promote senescence and cell death [37]. On the contrary, sphingosine-1-phosphate (S1P), the immediate product due to metabolism, fosters cell survival and proliferation [38]. Histologically confirmed glioma cells have shown a change from ceramides to S1P, leading to higher S1P concentrations than ceramide. With lower amounts of ceramides, apoptosis occurs less, which allows the glioma cells to spread more freely [38]. In addition, modification of ASAH1 in glioblastoma enables it to be secreted to interstitial tissues, allowing it to transfer their malignant potential to nearby cells [39].
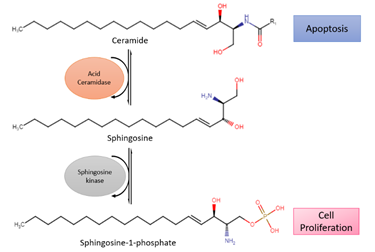
Figure 2. Reaction of acid ceramidase (ASAH1), a lysosomal enzyme that converts ceramides into sphingosine, which is further converted to sphingosine-1-phosphate (S1P) by sphingosine kinase. Ceramide promotes apoptosis while S1P stimulates cell survival and proliferation.
Previous studies have shown that glioblastomas express ASAH1 in high numbers. Doan et al. demonstrated that irradiated cell culture and tumor tissues have higher expression levels of ASAH1 compared to non-irradiated culture and tumor tissues, therefore leading to apoptotic resistance and glioblastoma recurrence [38]. This led to the identification of overexpression of ASAH1 as a potential biomarker associated with glioblastomas and the development of anticancer therapy. Although there are no drugs in clinical trials targeting ceramide signaling for glioblastomas, ASAH1 inhibitors (carmofur, N-oleoylethanolamine, and ARN14988) have been studied against multiple glioblastoma stem cell lines, U87, and patient-derived cell lines. In vitro studies of ASAH1 inhibitors have shown to be more effective against glioblastoma tumor cell lines compared to the Food and Drug Administration (FDA)-approved drug temozolomide (TMZ), therefore suggesting that ASAH1 inhibitors can restrain ASAH1 activity and increase tissue ceramide levels to induce apoptosis [40][41].
2.4. Vascular Endothelial Growth Factor (VEGF) Signaling Pathway
Vascular endothelial growth factor (VEGF), a potent angiogenic cytokine, stimulates the growth of new blood vessels to restore oxygen supply. The normal VEGF pathway starts when cells are lacking oxygen, which leads to the production of the hypoxia-inducible factor. This leads to releasing of VEGF followed by binding of the VEGF to VEGF receptors (VEGFRs), stimulating the tyrosine kinase pathway and ultimately resulting in angiogenesis. The normal signaling completes angiogenesis during embryonic development, collateral circulation, and following muscle injury and wounds [42].
Unfortunately, VEGF also plays a key role in promoting angiogenesis in glioma stem cells and optimizing the function and survival of its microenvironment. For survival of glioblastoma, a vascular supply must be maintained, and early extensions in the growing tumor receive this vascular supply by angiogenesis [43]. Hence, blocking the VEGF pathway and thereby inhibiting angiogenesis would be an effective strategy to treat the disease. Various anti-angiogenic agents have been shown to be effective in blocking the VEGF pathway, thereby treating several different cancers [44]. Though anti-VEGF therapy has been widely used and has shown benefits in the reduction of vasogenic edema associated with this disease, the overall survival benefit and resistance to therapy are yet to be improved. However, several approaches using combination therapy with radiotherapy, immunotherapy, cytotoxic drugs etc., in addition to anti-VEGF therapy showed improved results [45][46]. A recent study on combination therapy with platelet-derived growth factor (PDGF) inhibitors showed more promising results when combined with anti-VEGF therapy in terms of survival benefit and sensitization to therapy [47].
In the clinical setting, several receptor tyrosine kinase inhibitors (TKIs) such as tivozanib, cediranib, lenvatinib, sorafenib, sunitinib, and pazopanib are currently being studied for VEGFR inhibition. In addition, other therapeutic agents such as the TTAC-001 antibody, the VXM01 vaccine, and combination treatment with bevacizumab are also currently being studied. There are about 10 ongoing clinical trials and three recently published major clinical trials that are based on VEGF and VEGFR as the therapeutic targets for glioblastomas.
2.5. PDGF Signaling
Platelet-derived growth factor (PDGF) became a target for therapy for glioblastoma due to its ability to promote glioblastoma proliferation and survival [48]. In normal glial cells, PDGF signaling starts with the binding of the PDGF ligands such as PDGFA, PDGFB, and PDGFC to the platelet-derived growth factor receptor (PDGFRα or PDGFRβ). The PDGFR is classified as a cell surface receptor tyrosine kinase (RTK). Upon binding, the PDGFRs dimerize, allowing the subunits to cross phosphorylate several tyrosine residues in the receptor. This activated form acts as a docking site for multiple protein complexes to activate many signal transduction cascades, ultimately leading to DNA synthesis and cell proliferation [49][50].
On the contrary, a PDGF autocrine loop is exhibited in glioblastomas which should be absent in normal brain tissue [36]. Multiple observations have found PDGF overexpression in glioblastomas. PDGFA and PDGFB are highly expressed in comparison to the other ligands, with PDGFC being expressed the least [51]. Westermark noticed that the PDGFRα gene is amplified, mutated, or rearranged in glioblastoma tumors, playing a role in oncogenesis [52]. Similarly, Shih et al. found PDGF and PDGFR to be overexpressed in glial tumor cell lines and samples correlating with higher tumor grade. Autocrine signaling in tumor proliferation was tested in cell culture where PDGF inhibitors were able to limit colony activity and cell growth [49]. Popescu et al. investigated a PDGFR inhibitor, AG1433, and discovered that both the growth factor and its receptors can control cell proliferation, differentiation, and apoptosis in glioblastoma. They remarked that it was able to reduce cell survival to 56.5% with the highest concentration (100 μM) at 72 h [53]. Another study by Hong et al. found the TKI imatinib to be successful at enhancing the radiosensitivity and chemosensitivity of gliomas. Moreover, it has been observed that it can radiosensitize the cells and inhibit tyrosine phosphorylation of numerous intracellular proteins in a dose-dependent manner [54]. Another PDGFRα inhibition study conducted by Mangiola et al. found a significant decrease in cell proliferation in core cancer stem cells, by about 38 ± 9.5%. They also observed a decrease in the modulation of PDGFRα expression [55]. These studies indicate that PDGF is a well-studied pathway that could lead to possible treatments for glioblastoma.
In clinical settings, several TKIs such as tandutinib, crenolanib, sorafenib, sunitinib, and pazopanib are currently being studied. There are about six ongoing clinical trials and two recently published major clinical trials that are based on PDGF and PDGFR as the therapeutic targets for glioblastoma.
2.6. Epidermal Growth Factor Receptor (EGFR) Pathway
The epidermal growth factor receptor (EGFR) is a transmembrane cell RTK that binds extracellular signaling ligands such as epidermal growth factors and transforming growth factor-α to its extracellular domain. In normal glial cells, the EGFR pathway starts when the receptor binds to its signaling ligand and becomes activated, undergoing transitions to an active homodimer from an inactive monomer. This dimerization induces intracellular protein-tyrosine kinase activity and results in tyrosine residues being autophosphorylated in the C-terminal domain of EGFR. Such autophosphorylation stimulates the initiation of many signal transduction cascades, which ultimately lead to DNA synthesis, cell proliferation, migration, and adhesion [56].
Mutations in EGFR have been widely recognized to be involved in the pathogenesis of glioblastomas. The amplification of EGFR was found to be more commonly present in primary glioblastomas (40%), and rarely present in secondary glioblastomas [57]. Furthermore, EGFR amplification was found to be rare or nonexistent in pediatric glioblastomas [58]. In a population-based study conducted by Ohgaki et al., EGFR amplification was found to be detected only in glioblastoma patients older than 35 years, confirming the results of the previous study [59]. For tumors with amplified EGFR expression, about half of those cases have the EGFRvIII variant, which is an ideal target for therapies [60][61].
Though EGFR was one of the first molecule linked to oncogenesis of glioblastoma, targeting it has been challenging in this disease. Hence, recent studies have focused on both immunotherapy as well as tyrosine kinase inhibitors (TKIs). For example, OSI-774, an EGFR-TKI, has shown to be promising in a study conducted by Halatsch et al. They showed that it induces apoptosis in malignant glioblastoma and is a promising agent against secondary glioblastoma [62]. However, phase I/II clinical trials of another TKI, lapatinib, showed limited antitumor activity in patients. Though TKIs are promising, EGFR inhibitors in the pre-clinical settings as well as drug delivery and activity must be evaluated further [63]. EGFR-targeting therapeutic agents such as dacomitinib,nimotuzumab, ABBV-321, AMG596, CART-EGFRvIII T cells, EGFR(v)-EDV-DOX, axitinib, cabozantinib, neratinib, afatinib, alectinib, and tesevatinib are currently in clinical studies. From January 2017 to September 2019, about six major clinical trials were published, while currently there are about 19 ongoing clinical trials based on EGFR-targeting therapeutic agents and tyrosine kinase inhibition for glioblastomas.
2.7. PI3K/AKT/mTOR Pathway
The PI3K/AKT/mTOR pathway (Figure 3) is a vital intracellular signaling pathway for regulating the cell cycle. Phosphatidylinositol 3-kinases (PI3Ks) are intracellular signal transducer enzymes that can activate serine/threonine-specific protein kinase (AKT) through phosphorylation. Subsequently, AKT can activate the mammalian target of rapamycin (mTOR). mTOR forms two complexes which are characterized by different binding partners; mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [64]. mTORC1 is rapamycin-sensitive and is activated by at least five cues (growth factors, stress, energy status, oxygen, and amino acid concentration), and promotes glial cell growth upon activation by eukaryotic translation initiation factor 4E binding protein 1(E4BP1) and ribosomal protein S6 kinase (S6K) [64][65]. Conversely, mTORC2 is insensitive to rapamycin, which drives the glial cell proliferation, motility, and survival through the activation of AGC protein kinases [64][65][66]. However, it is found that overactivation of the PI3K/AKT/mTOR pathway reduces in the survival of glioblastoma patients and increases in the aggression of the tumor as it overstimulates processes responsible for cell proliferation, survival and migration in glioblastoma [67][68]. Therefore, researchers have identified PI3K, AKT, and mTOR as molecular targets for glioblastomas.
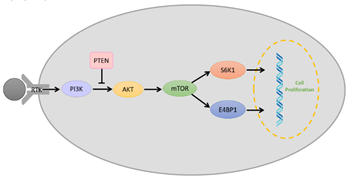
Figure 3. Schematic representation of a simplified overview on the PI3K/AKT/mTOR pathway with the role of phosphate and tensin homolog (PTEN) and receptor tyrosine kinase (RTK).
Recently, a few preclinical trials have found mTOR inhibitors to be successful. For example, Mecca et al. found that CC214-1 and CC214-2, mTOR kinase inhibitors, were capable of inhibiting glioblastoma growth by blocking mTOR2C2 activity both in vitro and in vivo. In the clinical setting, PI3K inhibitors such as BKM120, regorafenib, GDC-0084, and fimepinostat as well as mTOR inhibitors such as temsirolimus, everolimus, CC-115, ABI-009, AZD2014, sapanisertib, and siroquine are currently being studied. From January 2017 to September 2019, about four major clinical trials were published, while currently, there are about 11 ongoing clinical trials based on P13K and mTOR inhibition for glioblastomas.
2.8. Phosphate and Tensin Homolog (PTEN) Signaling
Another key element associated with glioblastoma in the PI3K pathway is phosphate and tensin homolog (PTEN). PTEN is a tumor suppressor that antagonizes PI3K signaling and prevents AKT activation via its lipid phosphatase activity (Figure 3) [69]. In glioblastomas, it has been reported that PTEN is inactivated due to mutations. A single mutation in one of the homolog genes is insufficient to initiate tumor growth; however, the deletion of one or both results in uncontrollable cell growth [70]. It has also been found that PTEN can sensitize glioma cells to chemotherapy and radiation therapy [71], hence making PTEN a molecular target for glioblastoma immunotherapy.
Recent developments in the preclinical setting have focused on correction to PTEN mutation. A study reported the correction of PTEN in glioblastoma using the adeno-associated virus-mediated gene that reduced the cellular proliferation in the glioblastoma cell lines, indicating that it could be a potential treatment for this disease [72]. Furthermore, another study illustrated that correction of the mutant allele of PTEN in glioblastoma cells lines (42MGBA and T98G) using gene editing resulted in reduced cell proliferation [73].
2.9. SHH Signaling
In normal glial cells, signaling starts with the sonic hedgehog (SHH) glycoprotein binding to and inactivating the protein Patched1 and co-receptors, leading to inactivation of the protein Smoothened (SMO). However, when SMO is activated, the nuclear localization of glioma-associated (GLI) transcription factors takes place. Once GLI enters the nucleus it leads to the activation of GLI1 and GLI2 transcription factors. Such activation promotes target activation in SHH signaling, leading to proliferation, angiogenesis, epithelial-to-mesenchymal transition, and stem cell self-renewal [74][75]. In glioblastomas, the abnormal activation of SHH signaling typically by mutation in Patched1 and/or activating mutations in SMO leads to the transformation of adult stem cells into glioblastoma stem cells.
Therefore, SHH signaling has become one of the focal points for glioblastoma treatment since mutations in the pathway play a key role in cell proliferation and tumorigenesis. Since SMO inhibition prevents downstream activation of GLI, SMO is an important molecular target for the development of SHH pathway inhibitors [76]. SMO inhibitors such as vismodegib, trametinib, and glasdegib have been under investigation for glioblastoma [77].
3. Conclusions
Tumor heterogeneity, patient-to-patient variability, and different stages of disease progression at the time of diagnosis foster complexity in the treatment of glioblastoma. While there are a few FDA-approved multimodal-approach treatments for glioblastoma, survival is still poor in majority of the patients. Understanding the molecular-level information on the mechanistics of neoplasms has led to the design of multiple new compounds which are now under investigation at different stages of clinical development. Based on the ongoing clinical trials, new treatment options are likely to evolve in the coming years. In addition, extensive research is ongoing to develop other novel strategies to better combat the disease. Ultimately, the overall goal is to lessen patient suffering by providing a better standard of life and increasing overall survival.
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018, 20, iv1–iv86, doi:10.1093/neuonc/noy131.
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.U. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed. Pharmacother. 2017, 92, 681–689, doi:10.1016/j.biopha.2017.05.125.
- Ferguson, S.; Lesniak, M.S. Percival Bailey and the classification of brain tumors. Neurosurg. Focus 2005, 18, e7, doi:10.3171/foc.2005.18.4.8.
- Zulch, K.J.; Wechsler, W. Pathology and Classification of Gliomas. In Progress in Neurological Surgery; Karger Publisher: Basel, Switzerland, 1968; Volume 2, pp. 1–84.
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435, doi:10.1111/bpa.12171.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820, doi:10.1007/s00401-016-1545-1.
- Huang, J.; Campian, J.L.; Gujar, A.D.; Tsien, C.; Ansstas, G.; Tran, D.D.; DeWees, T.A.; Lockhart, A.C.; Kim, A.H. Final results of a phase I dose-escalation, dose-expansion study of adding disulfiram with or without copper to adjuvant temozolomide for newly diagnosed glioblastoma. J. Neurooncol. 2018, 138, 105–111, doi:10.1007/s11060-018-2775-y.
- Cancer, I.A.f.R.o. WHO Classification of Tumours of the Central Nervous System; WTO: Geneva, Switzerland, 2016; Volume 1.
- Stöppler, M.C.; Shiel, W.C.; Credo Reference (Firm); WebMD (Firm). Webster's New World Medical Dictionary, 3rd ed.; redo Reference: Boston, MA, USA; Wiley: Hoboken, NJ, USA, 2014; p 1 (accessed on 10 March 2020).
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94, doi:10.1038/nrclinonc.2017.166.
- Yamaguchi, S.; Kobayashi, H.; Terasaka, S.; Ishii, N.; Ikeda, J.; Kanno, H.; Nishihara, H.; Tanaka, S.; Houkin, K. The impact of extent of resection and histological subtype on the outcome of adult patients with high-grade gliomas. Jpn. J. Clin. Oncol. 2012, 42, 270–277, doi:10.1093/jjco/hys016.
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro Oncol. 2002, 4, 278–299, doi:10.1093/neuonc/4.4.278.
- Preusser, M.; de Ribaupierre, S.; Wohrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21, doi:10.1002/ana.22425.
- Aoki, T.; Hashimoto, N.; Matsutani, M. Management of glioblastoma. Expert Opin. Pharmacother. 2007, 8, 3133–3146, doi:10.1517/14656566.8.18.3133.
- Sanai, N.; Berger, M.S. Recent surgical management of gliomas. Adv. Exp. Med. Biol. 2012, 746, 12–25, doi:10.1007/978-1-4614-3146-6_2.
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121, doi:10.3978/j.issn.2305-5839.2015.05.10.
- Ryken, T.C.; Frankel, B.; Julien, T.; Olson, J.J. Surgical management of newly diagnosed glioblastoma in adults: Role of cytoreductive surgery. J. Neurooncol. 2008, 89, 271–286, doi:10.1007/s11060-008-9614-5.
- Barbagallo, G.M.; Jenkinson, M.D.; Brodbelt, A.R. 'Recurrent' glioblastoma multiforme, when should we reoperate? Br. J. Neurosurg. 2008, 22, 452–455, doi:10.1080/02688690802182256.
- Cabrera, A.R.; Kirkpatrick, J.P.; Fiveash, J.B.; Shih, H.A.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract. Radiat. Oncol. 2016, 6, 217–225, doi:10.1016/j.prro.2016.03.007.
- Minniti, G.; Filippi, A.R.; Osti, M.F.; Ricardi, U. Radiation therapy for older patients with brain tumors. Radiat. Oncol. 2017, 12, 101, doi:10.1186/s13014-017-0841-9.
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in Radiotherapy for Glioblastoma. Front. Neurol. 2017, 8, 748, doi:10.3389/fneur.2017.00748.
- Corso, C.D.; Bindra, R.S.; Mehta, M.P. The role of radiation in treating glioblastoma: Here to stay. J. Neurooncol. 2017, 134, 479–485, doi:10.1007/s11060-016-2348-x.
- Fedoy, A.E.; Yang, N.; Martinez, A.; Leiros, H.K.; Steen, I.H. Structural and functional properties of isocitrate dehydrogenase from the psychrophilic bacterium Desulfotalea psychrophila reveal a cold-active enzyme with an unusual high thermal stability. J. Mol. Biol. 2007, 372, 130–149, doi:10.1016/j.jmb.2007.06.040.
- Kaminska, B.; Czapski, B.; Guzik, R.; Krol, S.K.; Gielniewski, B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 2019, 24, doi:10.3390/molecules24050968.
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812, doi:10.1126/science.1164382.
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345, doi:10.1007/s11910-013-0345-4.
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325, doi:10.1001/jamaneurol.2014.1205.
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305, doi:10.1021/acsmedchemlett.7b00421.
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630, doi:10.1126/science.1236062.
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506, doi:10.3389/fonc.2019.00506.
- Lino, M.M.; Merlo, A.; Boulay, J.L. Notch signaling in glioblastoma: A developmental drug target? BMC Med. 2010, 8, 72, doi:10.1186/1741-7015-8-72.
- Yan, D.; Hao, C.; Xiao-Feng, L.; Yu-Chen, L.; Yu-Bin, F.; Lei, Z. Molecular mechanism of Notch signaling with special emphasis on microRNAs: Implications for glioma. J. Cell. Physiol. 2018, 234, 158–170, doi:10.1002/jcp.26775.
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16, doi:10.1002/stem.254.
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers (Basel) 2019, 11, doi:10.3390/cancers11030292.
- Ying, M.; Wang, S.; Sang, Y.; Sun, P.; Lal, B.; Goodwin, C.R.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Laterra, J.; Xia, S. Regulation of glioblastoma stem cells by retinoic acid: Role for Notch pathway inhibition. Oncogene 2011, 30, 3454–3467, doi:10.1038/onc.2011.58.
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029, doi:10.1002/stem.429.
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65, doi:10.1038/nrc3398.
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940, doi:10.3892/or.2017.5855.
- Nguyen, H.S.; Awad, A.J.; Shabani, S.; Doan, N. Molecular Targeting of Acid Ceramidase in Glioblastoma: A Review of Its Role, Potential Treatment, and Challenges. Pharmaceutics 2018, 10, doi:10.3390/pharmaceutics10020045.
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674, doi:10.18632/oncotarget.22637.
- Doan, N.B.; Nguyen, H.S.; Montoure, A.; Al-Gizawiy, M.M.; Mueller, W.M.; Kurpad, S.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; et al. Acid ceramidase is a novel drug target for pediatric brain tumors. Oncotarget 2017, 8, 24753–24761, doi:10.18632/oncotarget.15800.
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82, doi:10.1016/j.pharmthera.2015.05.005.
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011, 13, 566–579, doi:10.1093/neuonc/nor039.
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171, doi:10.1159/000488087.
- Okuda, T.; Tasaki, T.; Nakata, S.; Yamashita, K.; Yoshioka, H.; Izumoto, S.; Kato, A.; Fujita, M. Efficacy of Combination Therapy with MET and VEGF Inhibitors for MET-overexpressing Glioblastoma. Anticancer Res. 2017, 37, 3871–3876, doi:10.21873/anticanres.11767.
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology (Williston Park) 2015, 29, 720–727.
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O'Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439, doi:10.1038/s41467-018-05982-z.
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61.
- Shih, A.H.; Holland, E.C. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006, 232, 139–147, doi:10.1016/j.canlet.2005.02.002.
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97, doi:10.1186/1478-811X-11-97.
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271, doi:10.1038/s41598-017-15045-w.
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Ups. J. Med. Sci. 2014, 119, 298–305, doi:10.3109/03009734.2014.970304.
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Tache, D.E.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the VEGF and PDGF signaling pathway in glioblastoma treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837.
- Hong, J.D.; Wang, X.; Peng, Y.P.; Peng, J.H.; Wang, J.; Dong, Y.P.; He, D.; Peng, Z.Z.; Tu, Q.S.; Sheng, L.F.; et al. Silencing platelet-derived growth factor receptor-beta enhances the radiosensitivity of C6 glioma cells in vitro and in vivo. Oncol. Lett. 2017, 14, 329–336, doi:10.3892/ol.2017.6143.
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Pierimarchi, P.; Paldino, E.; Casalbore, P.; Felsani, A.; Vescovi, A.L.; Maira, G.; Mangiola, A. PDGF receptor alpha inhibition induces apoptosis in glioblastoma cancer stem cells refractory to anti-Notch and anti-EGFR treatment. Mol. Cancer 2014, 13, 247, doi:10.1186/1476-4598-13-247.
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453, doi:10.2353/ajpath.2007.070011.
- Watanabe, K.; Tachibana, O.; Sata, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223; discussion 223–214.
- Kraus, J.A.; Felsberg, J.; Tonn, J.C.; Reifenberger, G.; Pietsch, T. Molecular genetic analysis of the TP53, PTEN, CDKN2A, EGFR, CDK4 and MDM2 tumour-associated genes in supratentorial primitive neuroectodermal tumours and glioblastomas of childhood. Neuropathol. Appl. Neurobiol. 2002, 28, 325–333.
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899, doi:10.1158/0008-5472.CAN-04-1337.
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735, doi:10.1007/s40263-017-0456-6.
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855, doi:10.1158/1078-0432.CCR-17-0890.
- Halatsch, M.E.; Gehrke, E.E.; Vougioukas, V.I.; Botefur, I.C.; A-Borhani, F.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533, doi:10.3171/jns.2004.100.3.0523.
- Reardon, D.A.; Groves, M.D.; Wen, P.Y.; Nabors, L.; Mikkelsen, T.; Rosenfeld, S.; Raizer, J.; Barriuso, J.; McLendon, R.E.; Suttle, A.B.; et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin. Cancer Res. 2013, 19, 900–908, doi:10.1158/1078-0432.CCR-12-1707.
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, doi:10.3390/ijms19082453.
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479, doi:10.1155/2018/9230479.
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11, doi:10.1186/s12964-018-0220-7.
- Mantamadiotis, T. Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer. Cancers (Basel) 2017, 9, doi:10.3390/cancers9060060.
- Lino, M.M.; Merlo, A. PI3Kinase signaling in glioblastoma. J. Neurooncol. 2011, 103, 417–427, doi:10.1007/s11060-010-0442-z.
- Janbazian, L.; Karamchandani, J.; Das, S. Mouse models of glioblastoma: Lessons learned and questions to be answered. J. Neurooncol. 2014, 118, 1–8, doi:10.1007/s11060-014-1401-x.
- Romano, C.; Schepis, C. PTEN gene: A model for genetic diseases in dermatology. ScientificWorldJournal 2012, 2012, 252457, doi:10.1100/2012/252457.
- Lester, A.; Rapkins, R.; Nixdorf, S.; Khasraw, M.; McDonald, K. Combining PARP inhibitors with radiation therapy for the treatment of glioblastoma: Is PTEN predictive of response? Clin. Transl. Oncol. 2017, 19, 273–278, doi:10.1007/s12094-016-1547-4.
- Valdes-Rives, S.A.; Casique-Aguirre, D.; German-Castelan, L.; Velasco-Velazquez, M.A.; Gonzalez-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747, doi:10.1155/2017/7403747.
- Hill, V.K.; Kim, J.S.; James, C.D.; Waldman, T. Correction of PTEN mutations in glioblastoma cell lines via AAV-mediated gene editing. PLoS ONE 2017, 12, e0176683, doi:10.1371/journal.pone.0176683.
- Liu, Y.; Liu, X.; Chen, L.; Du, W.; Cui, Y.; Piao, X.; Li, Y.; Jiang, C. Targeting glioma stem cells via the Hedgehog signaling pathway. Neuroimmunol. Neuroinflammation 2014, 1, 9, doi:10.4103/2347-8659.139715.
- Takezaki, T.; Hide, T.; Takanaga, H.; Nakamura, H.; Kuratsu, J.; Kondo, T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011, 102, 1306–1312, doi:10.1111/j.1349-7006.2011.01943.x.
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 2016, 8, doi:10.3390/cancers8020022.
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23, doi:10.1007/s11010-018-3448-z.

