 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tushar Janardan Pawar | -- | 4694 | 2025-04-08 19:04:52 | | | |
| 2 | Tushar Janardan Pawar | Meta information modification | 4694 | 2025-04-08 19:31:12 | | | | |
| 3 | Tushar Janardan Pawar | Meta information modification | 4694 | 2025-04-08 19:42:09 | | | | |
| 4 | Tushar Janardan Pawar | Meta information modification | 4694 | 2025-04-08 20:03:36 | | | | |
| 5 | Tushar Janardan Pawar | -4 word(s) | 4690 | 2025-04-08 20:18:21 | | | | |
| 6 | Tushar Janardan Pawar | -3 word(s) | 4687 | 2025-04-08 21:02:23 | | | | |
| 7 | Tushar Janardan Pawar | -63 word(s) | 4624 | 2025-04-09 04:04:44 | | | | |
| 8 | Tushar Janardan Pawar | -1 word(s) | 4623 | 2025-04-09 04:37:59 | | | | |
| 9 | Catherine Yang | Meta information modification | 4623 | 2025-04-09 04:58:27 | | | | |
| 10 | Catherine Yang | Meta information modification | 4623 | 2025-04-09 04:58:47 | | | | |
| 11 | Tushar Janardan Pawar | + 25 word(s) | 4648 | 2025-04-09 20:17:00 | | |
Video Upload Options
ApDOS (Aminocatalytic privileged Diversity-Oriented Synthesis) is a synthetic strategy that integrates the principles of aminocatalysis and diversity-oriented synthesis (DOS) to access structurally diverse libraries of privileged molecular scaffolds. Privileged structures—defined as frameworks capable of high-affinity binding to multiple biological targets—are key motifs in medicinal chemistry and chemical biology. ApDOS utilizes various aminocatalytic activation modes, including enamine, iminium ion, dienamine, trienamine, tetraenamine, and vinylogous iminium ion catalysis, to transform simple and readily available building blocks such as aldehydes and ketones into stereochemically rich and functionally elaborate architectures. The conceptual framework of ApDOS builds on the modularity and reactivity of aminocatalytic intermediates to populate biologically relevant regions of chemical space. By enabling mono- and multicomponent reactions—including cascade and domino sequences—ApDOS expands the synthetic utility of aminocatalysis beyond conventional target-oriented strategies. This methodology has proven particularly effective for the construction of spirocyclic, fused, bridged, and polycyclic scaffolds, often in a highly enantioselective and atom-economical manner. ApDOS represents a general platform for exploring molecular diversity via catalytic reactivity modes and offers significant potential for drug discovery and chemical probe development.
1. Introduction
Molecular diversity refers to the broad array of structurally varied chemical entities that occupy the extensive landscape of chemical space. This diversity underpins advances in contemporary disciplines such as chemical genetics and medicinal chemistry. Within the framework of chemical genetics, small molecules serve as probes capable of selectively interacting with one or multiple biological receptors, thereby modulating biomacromolecular functions and influencing biological phenotypes. These interactions enable the exploration of biological systems through reverse or forward chemical genetics strategies, where the latter is particularly valuable for concurrently uncovering bioactive structures and elucidating novel biological pathways.[1][2][3][4]
Despite the potential of chemical genetics in drug discovery, identifying effective lead compounds remains a considerable challenge. One widely adopted strategy involves the screening of libraries comprising structurally diverse small molecules, which enhances the likelihood of discovering potent biological modulators.[5][6][7][8] Nature has historically offered an abundant source of such molecular diversity through natural products—complex scaffolds that span wide regions of chemical space. While these molecules exhibit potent biological activities, their practical application as drug candidates is often constrained by limitations in abundance, isolation, and structural characterization.[9] Nonetheless, their unique frameworks have inspired the development of novel therapeutic agents via synthetic chemistry approaches.[10][11]
Historically, the synthesis of bioactive compounds has relied on Target-Oriented Synthesis (TOS), a method focused on the deliberate construction of specific molecular targets through retrosynthetic planning and convergent synthesis.[12] However, TOS often yields libraries with limited structural diversity, reducing the probability of identifying novel leads. To overcome this limitation, Diversity-Oriented Synthesis (DOS) was introduced as a strategy for efficiently generating libraries rich in structural complexity and diversity.[13][14][15][16][17][18] DOS employs a divergent synthetic approach, transforming simple precursors into a wide array of complex molecules through forward synthetic analysis, in contrast to the linear and focused pathways of TOS.
Since its introduction by Schreiber in 2000, DOS has spurred the development of numerous methodologies designed to produce molecular libraries featuring varied elements of diversity—such as appendages, functional groups, stereochemistry, and scaffolds.[19][20][21][22][23] These collections serve to explore underrepresented areas of chemical space and support the identification of innovative therapeutic candidates.
To further enhance the utility of DOS in drug discovery, the concept of privileged Diversity-Oriented Synthesis (pDOS) emerged. This strategy focuses on generating libraries centered on privileged structures—scaffold frameworks known to bind with high affinity to multiple biological targets.[24][25][26] By leveraging these frameworks, pDOS enables the rational design of compound libraries that are more likely to populate bioactive regions within chemical space.
Simultaneous to the evolution of DOS, the early 2000s also witnessed the resurgence of asymmetric catalysis, particularly via the use of small organic molecules—a development that marked the advent of organocatalysis.[27] The concept was simultaneously validated by the seminal contributions of David W. C. MacMillan[28] and Benjamin List,[29] both of whom introduced novel catalytic transformations in 2000. This breakthrough was later recognized with the 2021 Nobel Prize in Chemistry, highlighting the transformative impact of asymmetric organocatalysis on modern synthetic methodology.[30] Since then, organocatalysis has rapidly matured and is now recognized as the third fundamental strategy in asymmetric catalysis.[31] Among its various modalities, aminocatalysis has garnered particular attention due to its efficacy in stereoselective carbonyl functionalization, thereby granting access to a wide array of enantioenriched compounds.
Notably, the ApDOS concept (Aminocatalytic privileged Diversity-Oriented Synthesis) was introduced by Pawar et al. in 2018 as a unifying framework that leverages aminocatalytic activation modes to construct privileged molecular architectures from simple precursors.[32] The concept has since evolved, incorporating further developments such as the application of cross-conjugated systems, cascade strategies, and the use of polyenals and polyenones as advanced building blocks for generating sp³-rich frameworks.[33]
Recent advances in aminocatalysis, particularly in the use of photo- and dual-catalysis, machine-assisted reaction discovery, and biologically guided design, have further expanded the scope and utility of ApDOS approaches.[34][35][36][37][38] These emerging strategies are increasingly enabling the synthesis of structurally diverse small molecules suitable for phenotypic screening and functional chemical genomics.
2. Fundamental Activation Modes in Aminocatalysis: Framework for the ApDOS Concept
Aminocatalysis has emerged as a powerful strategy in asymmetric synthesis, primarily due to the ability of chiral primary and secondary amines to form transient covalent adducts with aldehydes and ketones. These reversible condensations yield highly organized intermediates that enable a variety of enantioselective transformations. The nature of the carbonyl substrate often dictates the activation pathway, thus allowing the design of rational, substrate-based methodologies. To date, approximately seven distinct activation modes have been elucidated, each serving as a foundational platform for numerous asymmetric transformations.[39][40][41][40][42]
At the core of these modes lie two fundamental principles of reactivity: HOMO-raising and LUMO-lowering. A canonical example of HOMO-raising is enamine activation, where enolizable carbonyl compounds, upon condensation with chiral amines, generate enamine intermediates. These species function as nucleophilic enolate equivalents capable of engaging in stereoselective C–C or C–heteroatom bond-forming reactions at the α-position of the carbonyl group.[40][41][40] Conversely, LUMO-lowering is exemplified by iminium ion activation. In this mode, α,β-unsaturated aldehydes or ketones react with chiral amines to form iminium ions, which facilitate asymmetric nucleophilic additions at the β-position, as well as pericyclic reactions.[42][43]
Building on these concepts, MacMillan and colleagues introduced the SOMO activation mode, wherein the enamine intermediate undergoes one-electron oxidation to generate a 3π-radical cation. This intermediate enables asymmetric radical transformations and has been foundational in merging aminocatalysis with photoredox catalysis, thus broadening the reaction landscape of the field.[44][45]
Beyond classical enamine and iminium ion pathways, the evolution of aminocatalysis has led to extended conjugated intermediates, such as dienamines,[46][47] linear trienamines,[39][48] cross trienamines,[49] tetraenamines,[39] and vinylogous iminium ions.[50][51] These extended systems allow selective functionalization at remote positions—typically five to seven bonds away from the catalyst’s site of interaction—thereby accessing previously uncharted chemical space.[52]
Parallel to the development of activation modes, significant progress has been made in catalyst design. Aminocatalysts based on pyrrolidine, imidazolidinone, and cinchona alkaloid scaffolds have been engineered not only to facilitate specific activation pathways but also to promote efficient stereocontrol. These catalysts often function through steric shielding or bifunctional hydrogen bonding, depending on their structural modifications and appended groups.
The collective success of aminocatalysis stems from the synergy between new activation concepts and innovative catalyst designs. Importantly, aminocatalytic transformations have enabled the synthesis of diverse molecular frameworks that map onto biologically relevant areas of chemical space. Many of these structures are considered privileged motifs, as they exhibit high affinity for multiple classes of biological targets.
Aminocatalysis and privileged Diversity-Oriented Synthesis (pDOS) share the common objective of generating structurally diverse molecules with high potential for biological activity. The intersection of these strategies has given rise to the concept of Aminocatalytic privileged Diversity-Oriented Synthesis (ApDOS).[32] This approach utilizes simple carbonyl compounds, such as aldehydes and ketones, as starting materials. In the presence of aminocatalysts, these substrates undergo one or more defined activation modes—such as enamine, iminium ion, or vinylogous intermediates—to form key chiral scaffolds.
These intermediate structures serve as platforms for subsequent diversification into privileged architectures through selective, often cascade-based, transformations. By leveraging the intrinsic reactivity of aminocatalytic intermediates and their compatibility with multiple reaction partners, ApDOS facilitates the efficient and stereoselective synthesis of molecular libraries tailored for applications in medicinal chemistry, chemical biology, and functional screening (see Figure 1).[32]
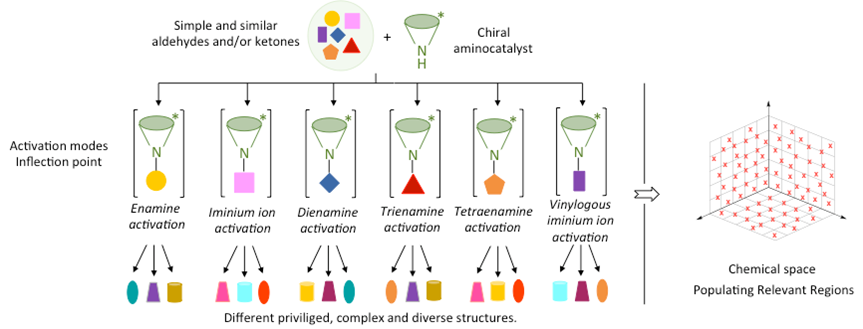
Figure 1. Aminocatalytic privileged Diversity-Oriented Synthesis concept.
3. ApDOS by Iminium Ion Catalysis
Among the foundational pathways in aminocatalysis, iminium ion activation has become a cornerstone for developing asymmetric reactions, particularly in the context of nucleophilic additions and pericyclic transformations. One of the earliest demonstrations of its potential was presented by MacMillan and co-workers, who showed that chiral iminium intermediates could effectively mediate enantioselective Diels–Alder reactions.[53] Since that landmark study, numerous methodologies have emerged that exploit this activation mode for the construction and diversification of optically active compounds of interest.
A particularly valuable application of iminium catalysis is in the asymmetric Michael addition of nucleophiles to α,β-unsaturated aldehydes, enabling selective C–C and C–heteroatom bond formation at the β-position. The broad scope of compatible nucleophiles allows for post-catalytic diversification through intra- or intermolecular transformations, often within a single reaction vessel. One representative example involves the one-pot synthesis of quinolizidines 3 via conjugate addition of electron-deficient amides 2 to enals 1, followed by a cyclocondensation and acid-catalyzed ring closure. This approach affords compounds 3a–g in high yields with excellent stereocontrol (Scheme 1a).[54]
Similarly, a multicomponent protocol for the assembly of octahydroacridine derivatives 7 was developed by combining alkenyl-malononitriles 5 with enals 4, followed by in situ Povarov cyclization via addition of anilines 6. This cascade sequence delivers the desired products with substantial molecular diversity due to variation in all three components (Scheme 1b).[55] Another illustrative case involves a three-component sequence encompassing oxa-Michael, Michael, and aldol steps, enabling the synthesis of chroman-fused spirooxindoles 11. This method generates libraries of biologically relevant molecules with moderate to high yields and excellent enantioselectivities (Scheme 1c).[56]
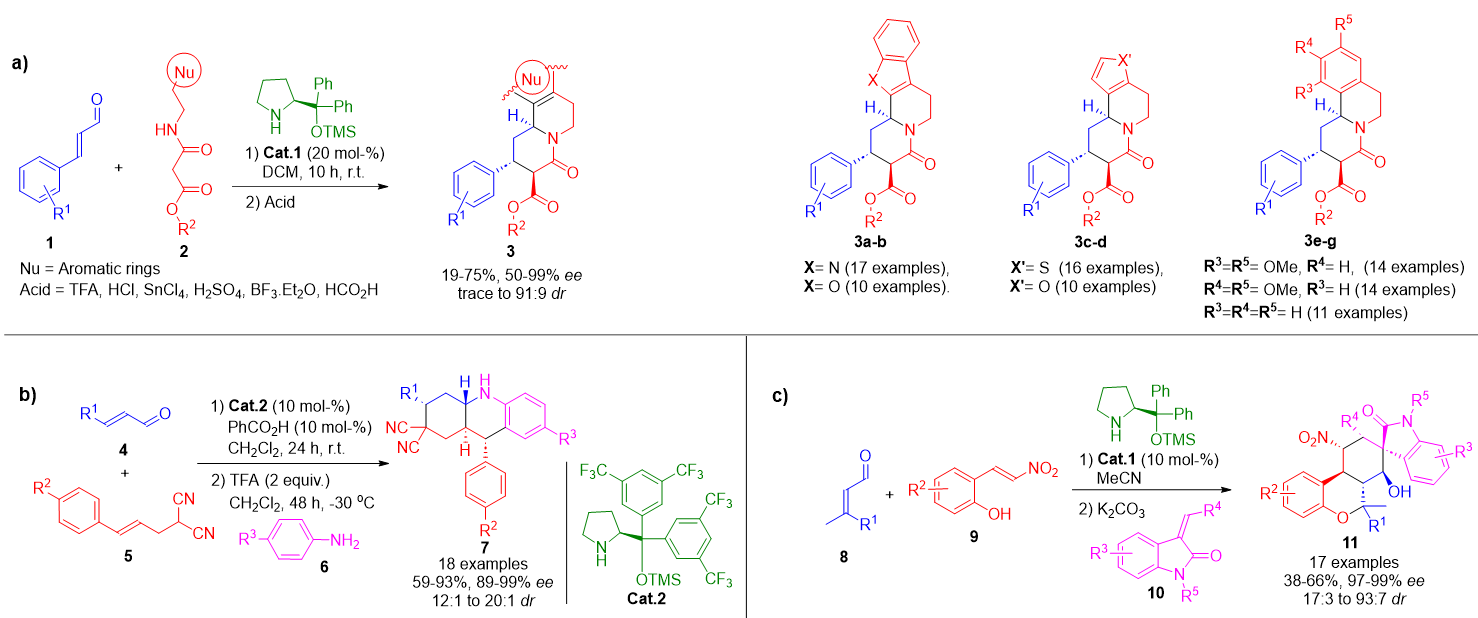
Scheme 1. a) One-pot aminocatalytic synthesis of enantioenriched quinolizidines via conjugate addition, condensation, and acid-promoted cyclization. b) Asymmetric multicomponent construction of octahydroacridines via a sequential Michael/Povarov reaction. c) Modular synthesis of chroman-fused spirooxindoles using a cascade oxa-Michael/Michael/aldol approach.
Five-membered heterocycles remain a central motif in medicinal and natural product chemistry, and [3+2] cycloadditions have proven particularly effective in accessing these scaffolds. Iminium ion intermediates, formed from α,β-unsaturated aldehydes and chiral amines, have served as activated dipolarophiles in diverse annulation strategies. Over the past two decades, numerous reports have documented the enantioselective synthesis of isoxazolidines via reactions between enals and nitrones as 1,3-dipoles.[57]
In a more recent variation, a structurally modified nitrone 12 bearing an electron-withdrawing group underwent aminocatalytic [3+2] annulation to yield N-hydroxypyrrolidines 13—instead of the expected isoxazolidines—with high enantiocontrol and moderate to excellent yields (Scheme 2a).[58] Another significant advance involves the use of dihydroisoquinolinium ylides, generated in situ, which participated in a cycloaddition with iminium-activated enals to afford hexahydropyrrolo-isoquinolines 17 with excellent optical purity (Scheme 2b).[59] Additionally, azomethine imines 19a were employed as dipolar species in a novel [3+2] cycloaddition. Depending on the conditions, the α,β-unsaturated aldehyde 18 could form either iminium or dienamine intermediates, providing divergent access to tetrahydroisoquinolines 20a with high chemo- and stereoselectivity (Scheme 2c).[60]
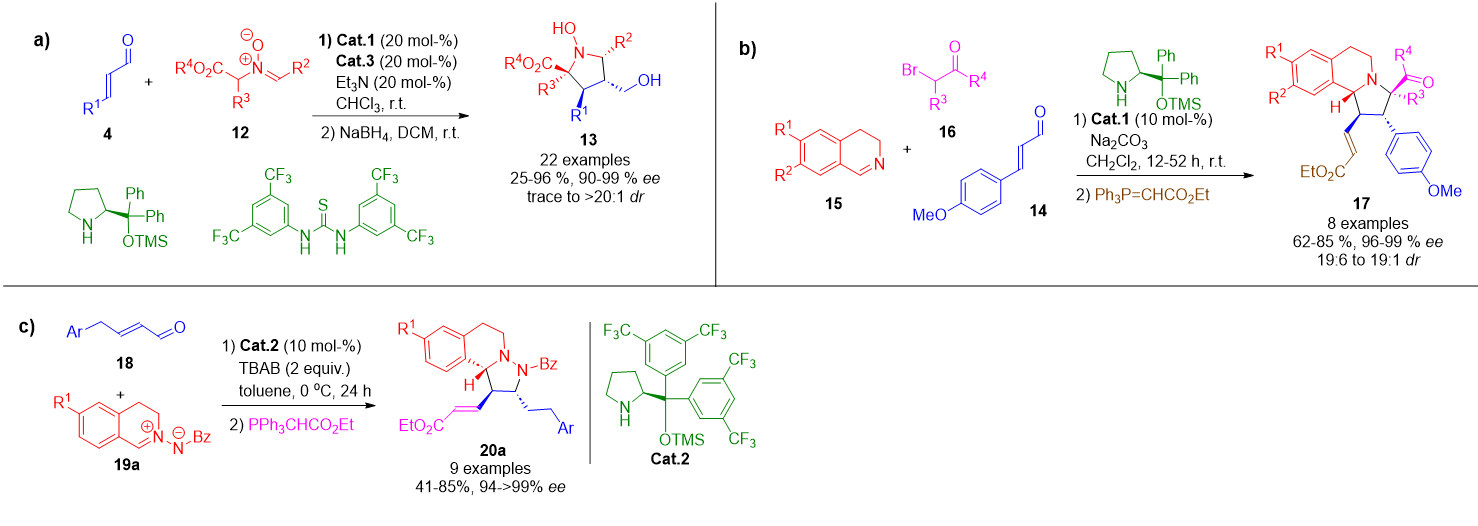
Scheme 2. a) Aminocatalytic [3+2] annulation of nitrones with enals under cooperative hydrogen-bonding and iminium ion activation, yielding N-hydroxypyrrolidines. b) Multicomponent cycloaddition involving dihydroisoquinolinium ylides and iminium-activated enals to access hexahydropyrrolo-isoquinolines. c) Divergent [3+2] cycloaddition of α,β-unsaturated aldehydes with azomethine imines, enabled by selective iminium or dienamine activation modes.
4. ApDOS by Enamine Catalysis
The earliest example of enamine catalysis in asymmetric synthesis dates back to 1971, when research teams at Hoffmann-La Roche (Hajos and Parrish)[61][62] and Schering AG (Eder, Sauer, and Weichert)[63][64] independently reported the intramolecular aldol condensation of achiral diketones using L-proline as a catalyst.[65] This transformation proceeds via enamine intermediate formation, enabling access to optically active bicyclic ketones. Although the formal conceptualization of organocatalysis occurred decades later, these early discoveries laid the foundation for what is now recognized as the field of asymmetric organocatalysis. The resulting Weiland–Miescher ketone, a key product of this reaction, continues to serve as a versatile building block in the total synthesis of sesquiterpenoids, diterpenoids, and steroidal frameworks.[66]
Over the past two decades, the enamine activation mode has been broadly developed for asymmetric catalysis, particularly in Michael additions involving electron-deficient olefins. These reactions frequently proceed through cascade or tandem sequences, leading to structurally diverse molecules with high stereocontrol.[15c–j] A representative application involves the transformation of N-protected 1-aminomethyl or 1-hydroxymethyl nitroolefins 22 with in situ generated enamines. This reaction yields heterocycles 23 featuring six-membered N- or O-centered rings and bearing up to four contiguous stereocenters (Scheme 3a).[67]
In another example, oxindole derivatives 24, possessing electron-withdrawing groups, serve as Michael acceptors in a multicomponent enamine-catalyzed process. The addition of aldehydes 21 subsequently initiates aldol and hemiacetalization steps. Final reduction of the intermediate yields spirocyclic oxindoles 25, fused with tetrahydropyran moieties, in good overall yields and excellent stereoselectivities (Scheme 3b).[68]
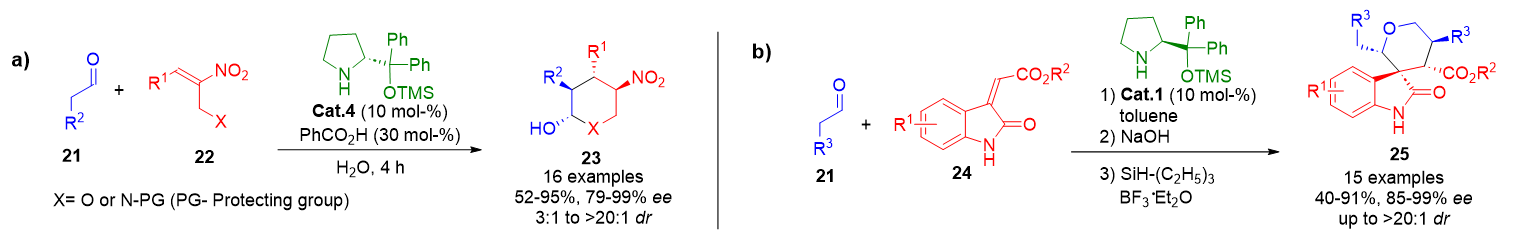
Scheme 3. a) Enamine-catalyzed Michael addition of aldehydes to nitroolefins followed by aminalization, yielding heterocycles with four stereogenic centers. b) Modular synthesis of spiro-oxindole derivatives via a Michael/aldol/hemiacetalization cascade.
A recently developed enamine-based method for generating aza-quaternary centers in alkaloids relies on the intramolecular acyl-Mannich cyclization. This sequence is initiated by condensation between a tethered acetal and an aldehyde to form an N,O-hemiaminal, which rearranges into an N-acyl iminium ion. Subsequent cyclization yields izidine alkaloid derivatives 27 with high diastereo- and enantioselectivity, across a range of five-, six-, and seven-membered ring systems (Scheme 4).[69]
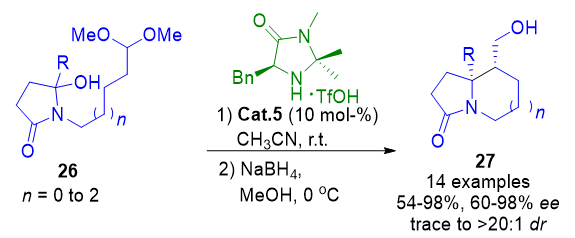
Scheme 4. Asymmetric synthesis of aza-quaternary centers via intramolecular acyl-Mannich cyclization of N,O-hemiaminals and tethered aldehydes.
Inverse-electron-demand (IED) Diels–Alder reactions have proven highly effective in constructing complex frameworks. In this context, enamines act as electron-rich dienophiles and have been successfully applied in IED Diels–Alder strategies, expanding the chemical space accessible through organocatalysis.
A prominent example involves the reaction of γ-lactam-derived cyclic enones 28 with enamine intermediates to form bicyclic dihydropyrans 29 with high levels of stereoselectivity (Scheme 5a).[70] Extending this concept, (Z)-2-ylideneoxindoles 30 function as electron-deficient heterodienes in an IED oxa-Diels–Alder reaction. This strategy enables the preparation of hydropyrano[3,2-b]indoles 31 with excellent functional group diversity and stereocontrol (Scheme 5b).[71]
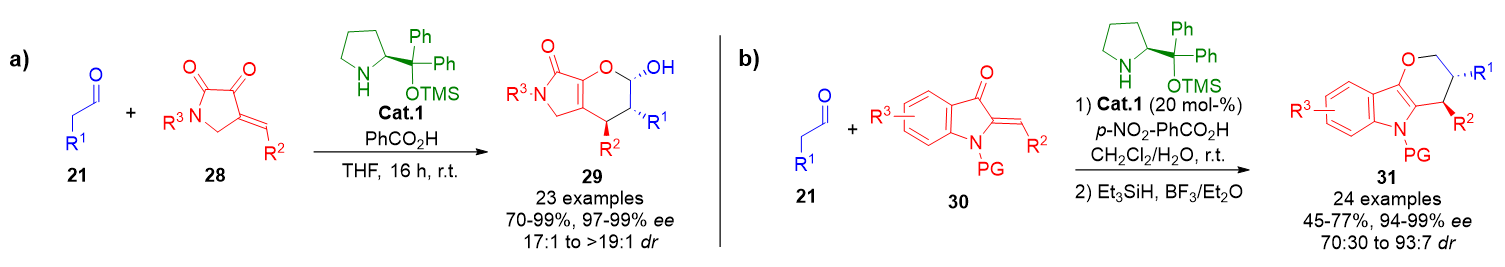
Scheme 5. a) Asymmetric IEDDA reaction of saturated aldehydes and γ-lactam-derived cyclic enones to form fused dihydropyrans. b) Enamine-catalyzed IEDDA reaction between saturated aldehydes and oxindole-derived heterodienes to generate hydropyrano[3,2-b]indoles.
Further advancing the ApDOS approach, an enamine-based aza-Diels–Alder reaction was developed involving N-sulfonyl-1-aza-1,3-butadienes 33 and aldehydes 32. The enamines formed in situ from the aldehydes undergo cycloaddition with the azadienes, followed by an intramolecular Friedel–Crafts reaction. This cascade sequence furnishes piperidine derivatives 34 with high stereochemical purity and molecular complexity (Scheme 6).[72]
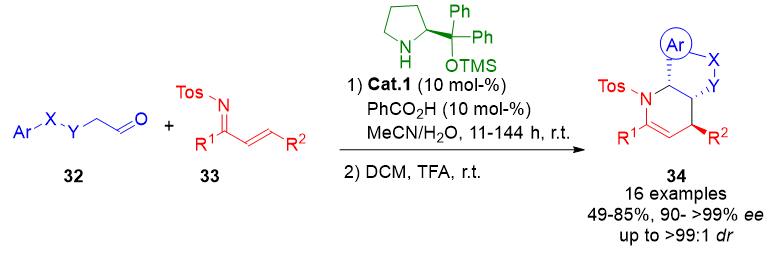
Scheme 6. Aminocatalytic IED aza-Diels–Alder reaction between N-sulfonyl-1-aza-1,3-butadienes and aldehyde-derived enamines, followed by Friedel–Crafts cyclization to access diverse piperidine scaffolds.
5. ApDOS by Dienamine Catalysis
As the exploration of aminocatalytic activation modes progressed, it became evident that the HOMO-raising principle could be extended beyond enamine intermediates. This led to the development of dienamine catalysis, wherein conjugated carbonyl compounds react with amines to form reactive dienamine species. These intermediates enable new stereoselective transformations, notably Diels–Alder reactions and cycloadditions, thus enriching the synthetic utility of ApDOS.
Among the privileged scaffolds accessible by dienamine catalysis, chromane derivatives are of particular significance due to their widespread occurrence in bioactive natural products. In 2012, Vicario et al. reported the construction of isochromane compounds 37 using a Diels–Alder/elimination cascade. This transformation employed racemic 5-acyloxydihydropyranones 35 as dienes and enolizable α,β-unsaturated aldehydes 36 as dienophiles, affording the products in high yields and excellent enantioselectivity (Scheme 7a).[73]
More recently, Albrecht and Bojanowski developed a related transformation involving chromene carboxylic acids 39 and enals 38 under dienamine catalysis. The process afforded a series of optically active dihydroxanthones 40, with enantiomeric excesses reaching up to 94%, thus exemplifying a robust diversification strategy (Scheme 7b).[74]
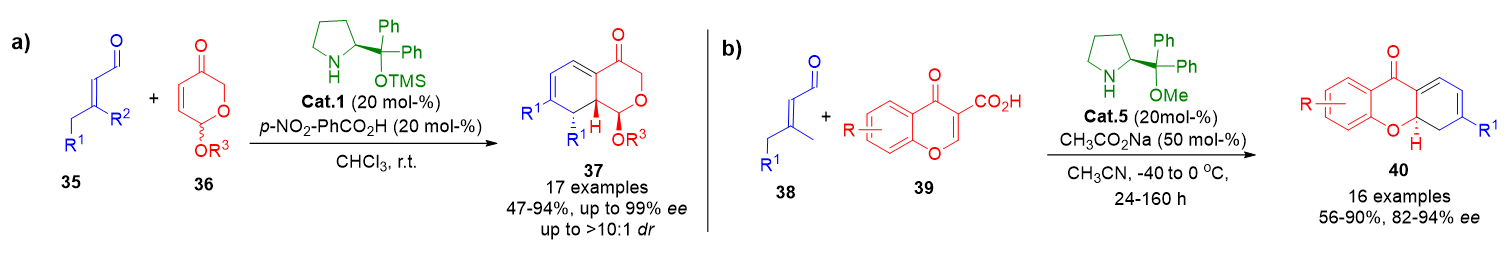
Scheme 7. a) Stereoselective Diels–Alder/elimination sequence catalyzed by dienamines for the synthesis of isochromane scaffolds. b) Dienamine-mediated diversification of chromene carboxylic acids to access 4,4a-dihydroxanthone derivatives.
Steroidal compounds constitute another class of privileged structures targeted by dienamine catalysis. In 2014, Jørgensen et al. developed a highly efficient one-step transformation for the synthesis of enantioenriched 14β-steroids 43 by reacting α,β-unsaturated aldehydes 41 with cyclic dienophiles 39. The process furnished the steroidal frameworks in high yields with enantiomeric excesses exceeding 99% (Scheme 8).[75] These products are structurally related to cardenolides and estrone-like molecules, highlighting their pharmaceutical relevance.
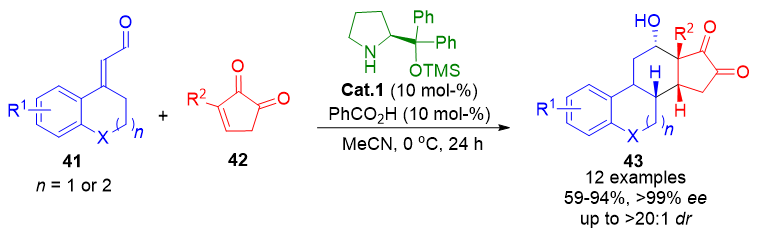
Scheme 8. Highly enantioselective one-step synthesis of 14β-steroids via dienamine-catalyzed Diels–Alder reactions with cyclic dienophiles.
A further application of dienamine catalysis involves the desymmetrization of cyclohexadienones through intramolecular [4+2] cycloadditions, yielding complex tricyclic frameworks 45. This method supports the incorporation of diverse substituents and delivers excellent stereocontrol (Scheme 9).[76] The resulting scaffolds are present in natural products such as momilactone A, azadirachtin, nagilactone, and other tricyclic terpenoid analogues.
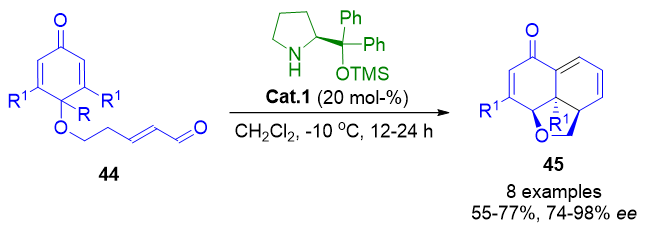
Scheme 9. Intramolecular dienamine-mediated Diels–Alder/aldol cascade for constructing optically active tricyclic architectures.
A dual activation mode was reported by García Ruano et al., who demonstrated that β-alkyl enals could react through either iminium ion or dienamine intermediates. In the presence of Cat.1, the dienamine pathway was favored, facilitating a [3+2] cycloaddition with azomethine imines, and leading to tetrahydroisoquinoline derivatives 20b in high yields and stereoselectivity (Scheme 10).[27] This bifunctional approach allows access to complex polycyclic structures via ApDOS using tunable activation strategies.
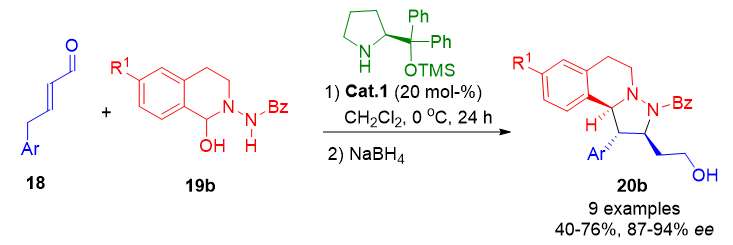
Scheme 10. Selective formation of tetrahydroisoquinoline scaffolds through dienamine-catalyzed [3+2] cycloadditions with azomethine imines.
While aldehydes are common precursors in enamine and dienamine chemistry, α-enolizable α,β-unsaturated ketones 46 have also been successfully employed to form cross-dienamine intermediates. Jørgensen et al. exploited this strategy to access norcamphor-like bicyclic scaffolds 48 from unsaturated ketones and dienophiles in high yields and excellent enantioselectivity (Scheme 11).[77] The resulting bicyclo[2.2.1]heptane frameworks are found in natural and synthetic molecules with diverse biological activities, including antiviral, antidiabetic, and antifungal properties.
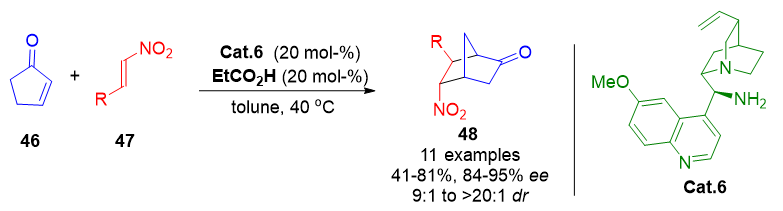
Scheme 11. Enantioselective synthesis of multifunctionalized norcamphor scaffolds via cross-dienamine-catalyzed Diels–Alder cycloadditions.
6. ApDOS by Trienamine Catalysis
A substantial portion of chemical space is occupied by cyclic and polycyclic frameworks, especially heterocycles with diverse substitution patterns. Among the most powerful tools for constructing such scaffolds are cycloaddition reactions, including their formal variants. In this context, trienamine catalysis, an extension of the aminocatalytic HOMO-raising concept, enables formal [4+2] cycloadditions and offers a strategic entry into enantioenriched cyclohexene derivatives.
The first demonstration of this activation mode appeared in 2011, where the combination of a chiral amine (Cat.7) with dienals 49 led to in situ formation of trienamines, which underwent stereoselective Diels–Alder-type reactions with various dienophiles 50. This stepwise process begins with the distal alkene of the trienamine (in an s-cis conformation) attacking the dienophile, forming a zwitterionic intermediate that collapses via 1,4-addition to give cyclohexenes 51 bearing all-carbon quaternary centers in high yield and enantioselectivity (Scheme 12a).[78] The strategy has since been broadened to include a range of carbon-based dienophiles. For instance, trisubstituted nitroolefins 53 have been employed to synthesize spirocyclohexene oxetanes 54 with high stereocontrol (Scheme 12b).[79]
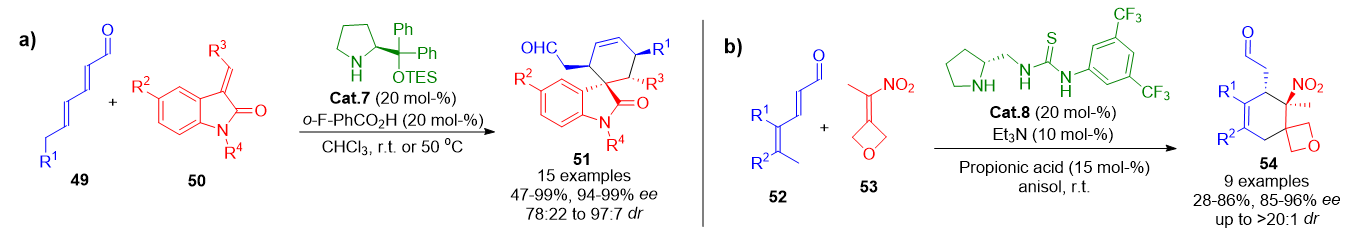
Scheme 12. a) Initial example of asymmetric trienamine-mediated Diels–Alder cycloadditions producing quaternary cyclohexenes. b) Stereoselective annulation of dienals and nitroolefins affording spiro-oxetane-cyclohexenes.
Heterocyclic scaffolds play a central role in medicinal chemistry, and hetero-Diels–Alder reactions remain indispensable for assembling six-membered heterocycles. Trienamine catalysis has been adapted to include heteroatom-centered dienophiles, such as dithioesters 56, which react to afford sulfur-containing heterocycles 57 with excellent enantiocontrol (Scheme 13a).[79] In another approach, dienones 58 were used as heterodienes in reactions with electron-deficient partners 59, catalyzed by Cat.9, leading to the formation of aza-heterocycles 60 with good yields and selectivity (Scheme 13b).[80]
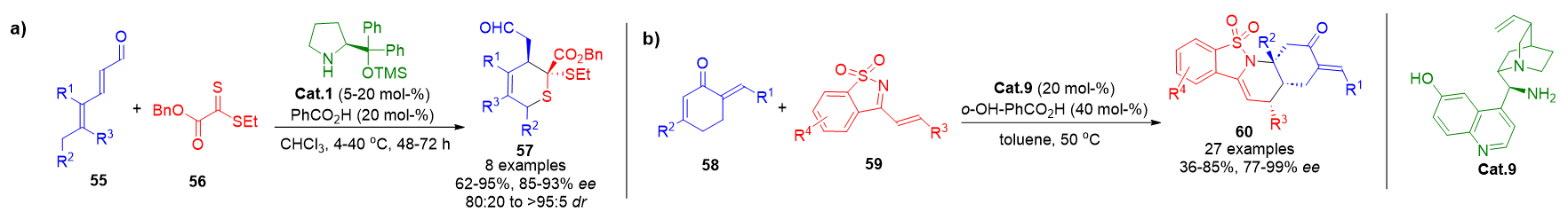
Scheme 13. a) Thio-Diels–Alder annulations using trienamine activation with dithioester dienophiles. b) Cross-conjugated trienamine-enabled aza-Diels–Alder cycloaddition with dienones.
The indole framework, ubiquitous in natural products and pharmaceutical compounds, has also been incorporated into ApDOS via trienamine catalysis. In one method, substrates 61 underwent de-aromatization to form ortho-quinodimethane-type intermediates, which then reacted with dienophiles 50 to furnish fused indole derivatives 62 with high enantioselectivity (Scheme 14a).[81] An alternative strategy used 3-nitroindoles 63 as indolyne equivalents. These species engaged in formal Diels–Alder reactions with dienals 55 via trienamine intermediates, forming re-aromatized cycloadducts 64 after elimination (Scheme 14b).[82]
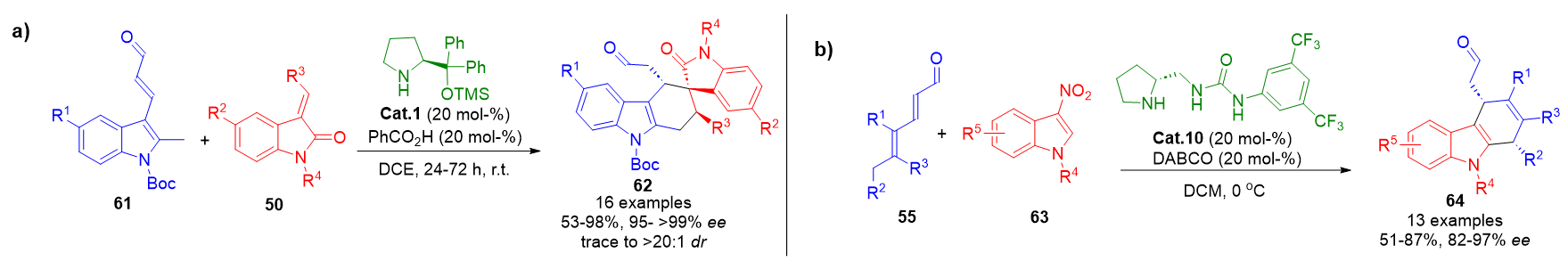
Scheme 14. a) Synthesis of enantioenriched fused indoles via trienamine-triggered de-aromatization and cycloaddition. b) Formal Diels–Alder annulation between dienals and 3-nitroindoles with subsequent re-aromatization.
Trienamine catalysis has also facilitated the assembly of polycyclic structures via cascade annulation strategies. The aldehyde group in the formal Diels–Alder adducts often enables secondary cyclizations, especially when dienophiles bear nucleophilic functionalities. Two notable examples include the synthesis of hydroisochromenes 66 and benzo[de]quinolones 69 through sequential [4+2] cycloaddition and O- or N-nucleophile ring closure, respectively (Scheme 15).[83][84]
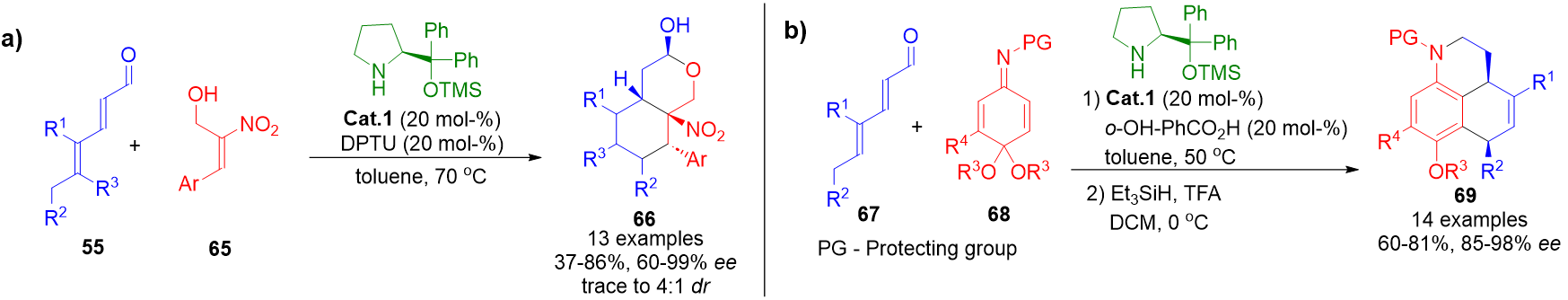
Scheme 15. a) Formation of hydroisochromenes via trienamine-mediated Diels–Alder/nucleophilic cyclization cascade. b) Synthesis of benzo[de]quinolones via Diels–Alder/aromatization/hemiaminal formation sequence.
In addition to fused polycycles, bridged bicyclic frameworks have been synthesized using cross-trienamine catalysis. This was illustrated by reacting cyclic dienals 70 with diverse dienophiles, yielding bicyclic products 71 with high enantiocontrol (Scheme 16).[49] This approach expands ApDOS to include rigid polycyclic motifs of biological relevance.
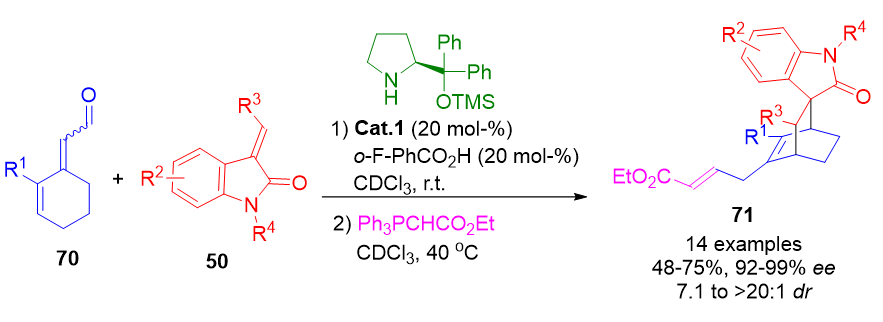
Scheme 16. Cross-trienamine catalysis enabling the asymmetric synthesis of enantioenriched bridged bicyclic compounds from cyclic dienals.
Although trienamine catalysis has predominantly employed aldehyde precursors, recent advances show that α,β-unsaturated ketones 46 can also generate cross-trienamine intermediates. This broadens substrate scope and enhances molecular complexity. For example, dienones reacted via trienamine catalysis with both cyclic dienones and 3-olefinic-7-azaoxindoles, delivering highly functionalized products 73 and 76 in good yields and stereoselectivity (Scheme 17).[85][86]
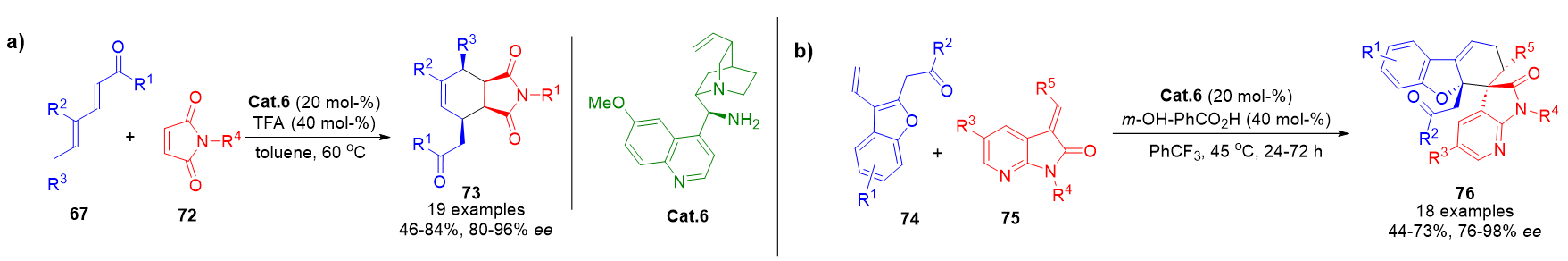
Scheme 17. a) Trienamine-mediated Diels–Alder cycloaddition between ketone-derived dienones yielding polycyclic products. b) Synthesis of azaoxindole derivatives via ketone-based trienamine activation.
Since the introduction of trienamine catalysis within the ApDOS framework, numerous innovative methodologies have been reported, further expanding the synthetic utility and structural diversity accessible through this activation mode.[87][88][89]
7. ApDOS by Tetraenamine Catalysis
Building upon the progression from enamine to dienamine and trienamine activation modes, the concept of tetraenamine catalysis has emerged as a further extension of the HOMO-raising strategy in aminocatalysis. This advanced activation mode leverages extended π-conjugation in carbonyl compounds, enabling transformations that are otherwise difficult to access. Although challenges such as reduced reactivity and complex regioselectivity accompany these systems, significant synthetic advances have been reported using carefully engineered substrates.
In one approach, cyclic trienals 77 were combined with olefinic oxindoles 78 under aminocatalytic conditions, furnishing spirooxindoles 79 with high stereoselectivity and moderate to good yields. The aldehyde moiety remained fixed across reactions, while structural diversity was introduced via varying substituents on the oxindole component (Scheme 18a).[90]
A related strategy involved linear trienals 80, where regioselectivity was finely tuned through strategic substitution patterns within the trienal framework. This led to the formation of spirooxindoles 82 in similarly favorable yields and enantiomeric excess (Scheme 18b).[53] Both methodologies also proved compatible with benzofuranones as alternative electrophilic partners, expanding the range of accessible scaffolds.[91]
In a further refinement, 5-allyl-substituted furfural derivatives 83 were utilized as substrates to generate tetraenamine intermediates via dearomatization in the presence of Cat.11. These intermediates underwent an inverse-electron-demand (IED) oxa-Diels–Alder reaction at a remote double bond, delivering spirooxindoles 85 with promising selectivity and synthetic efficiency (Scheme 18c).[92]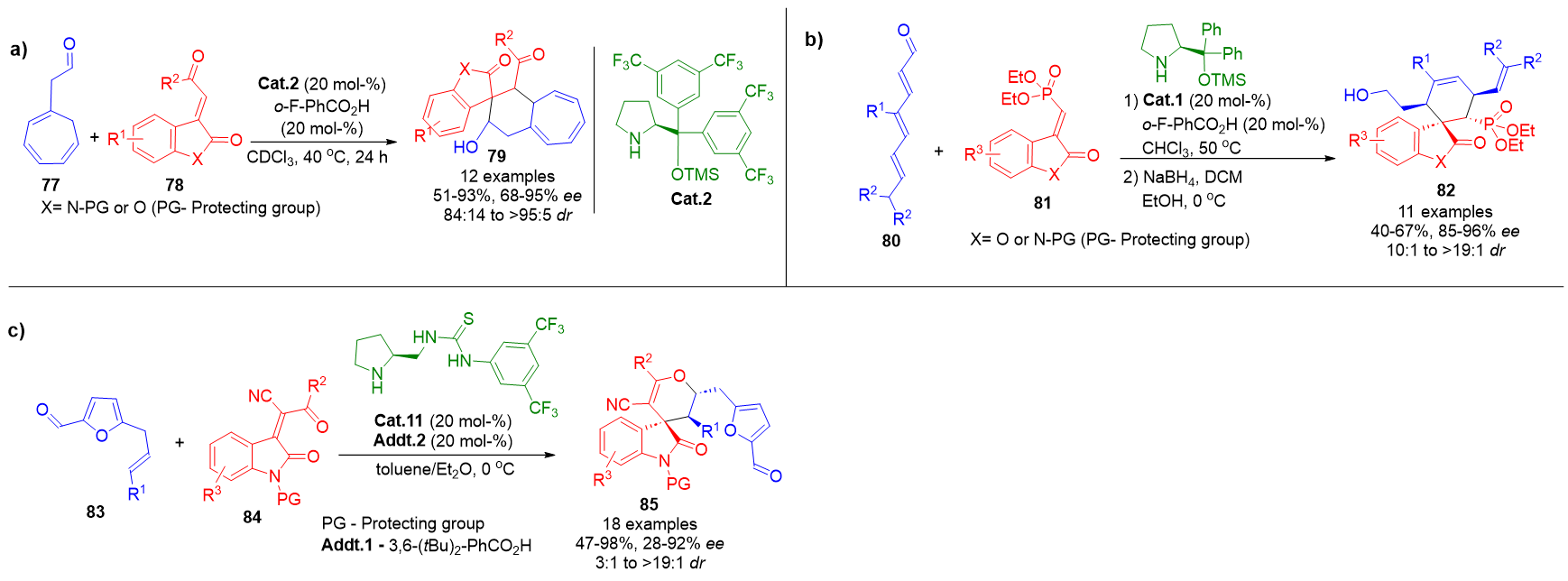
Scheme 18. a) Formation of spirooxindoles via tetraenamine-mediated cycloaddition using cyclic trienals and olefinic oxindoles. b) Stereoselective annulation of substituted linear trienals with oxindoles through tetraenamine activation. c) IED oxa-Diels–Alder reaction of dearomatized furfural derivatives enabling access to spirooxindole frameworks.
8. ApDOS by Vinylogous Iminium Ion Catalysis
In contrast to the HOMO-raising activation pathways, vinylogous iminium ion catalysis operates under the LUMO-lowering paradigm of aminocatalysis. This strategy combines classical iminium ion activation with the principle of vinylogy, enabling remote electrophilic activation across conjugated unsaturated aldehydes. The result is a powerful tool for regioselective, stereocontrolled transformations in complex molecule construction.
A notable early application was reported by Melchiorre et al., who designed a cascade reaction involving cyclic dienones 86 and 3-substituted oxindoles 89. The reaction proceeded through vinylogous iminium activation, followed by intramolecular aldol condensation, yielding spirocyclopentane oxindoles 88 in excellent yields and stereocontrol (Scheme 19a).[93]
In a complementary methodology, the same research group employed linear 2,4-dienals 90 to achieve a 1,6-addition with oxindoles 89, culminating in the formation of tetrahydrofuran spirooxindoles 91 via vinylogous iminium intermediates (Scheme 19b).[94]
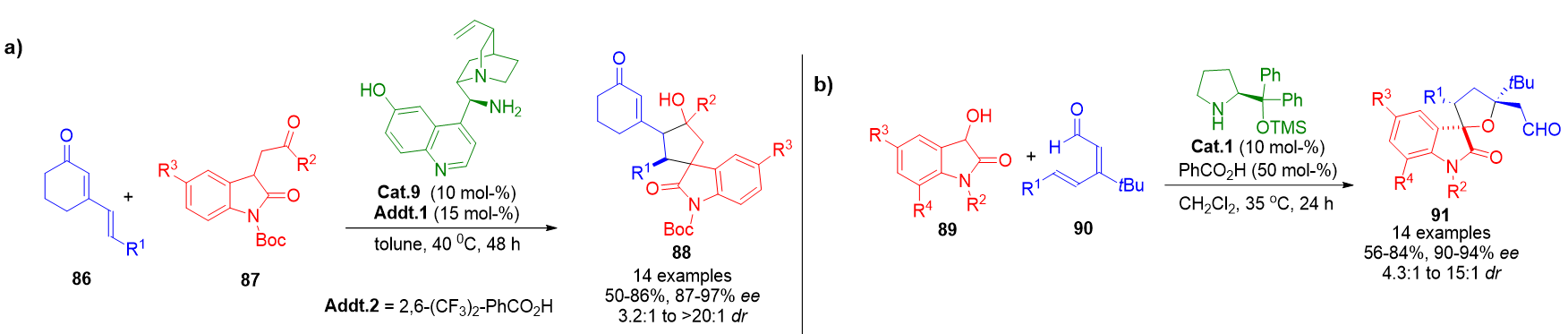
Scheme 19. a) Enantioselective synthesis of spirocyclopentane oxindoles via vinylogous iminium activation and intramolecular aldol condensation. b) Aminocatalytic 1,6-addition of oxindoles to dienals forming tetrahydrofuran-fused spirooxindole scaffolds.
The vinylogous strategy has also been extended to complex cascade 1,3-dipolar cycloadditions. Drawing inspiration from classical iminium ion-mediated isoxazolidine synthesis, 2,4-dienals 93 and 2,4,6-trienals 94 were subjected to sequential nitrone additions. These multistep processes generated bi- and tri-isoxazolidine products 95a and 95b containing six and nine stereogenic centers, respectively, via vinylogous and bis-vinylogous iminium intermediates (Scheme 20).[95] These cascades offer rapid access to densely functionalized molecular architectures from simple starting materials.
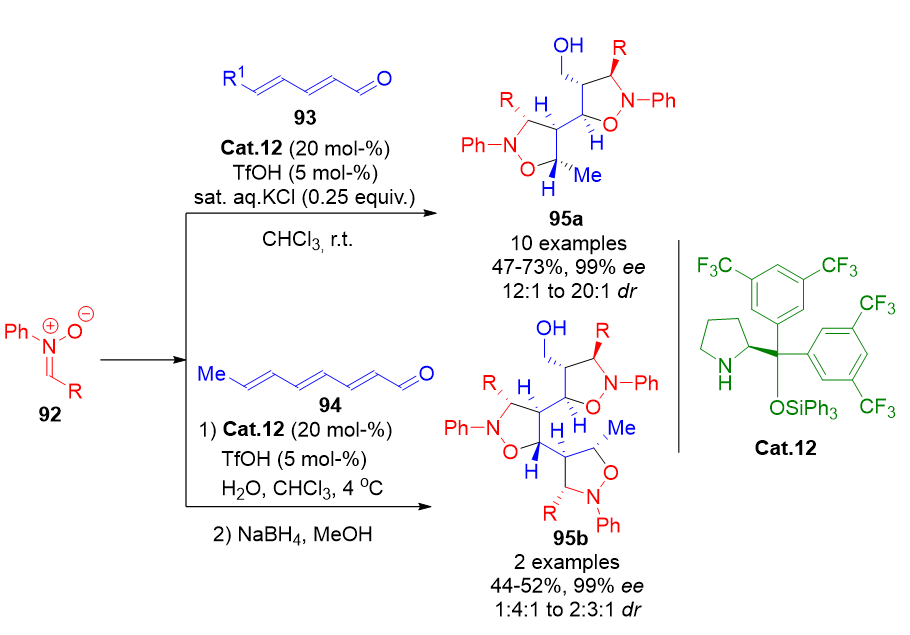
Scheme 20. Sequential 1,3-dipolar cycloaddition cascades via vinylogous and bis-vinylogous iminium ion intermediates for the stereoselective synthesis of bi- and tri-isoxazolidines.
9. ApDOS by Aminocatalytic Cascades
Aminocatalytic cascades represent a versatile strategy within the ApDOS framework, enabling the sequential orchestration of multiple activation modes to construct structurally complex and stereochemically rich molecular architectures. These transformations can integrate several components in a single process, offering a direct route to privileged scaffolds with high chemo-, regio-, and stereoselectivity. The strategic combination of multiple aminocatalytic pathways—such as enamine, iminium, dienamine, or their vinylogous analogs—facilitates efficient diversification of chemical libraries from simple precursors.
The archetypal design of such cascades often involves a sequential iminium–enamine mechanism, wherein the initial iminium ion formation promotes nucleophilic addition, followed by the generation of an enamine intermediate that engages an electrophilic partner. This dual-activation mode can occur within a single nucleophile bearing both reactive centers. A prominent example is the synthesis of spiro-tetrahydrothiopyran-oxindoles 98, accessed via a two-step cascade involving sequential carbon–carbon bond formation under asymmetric control (Scheme 21).[96]
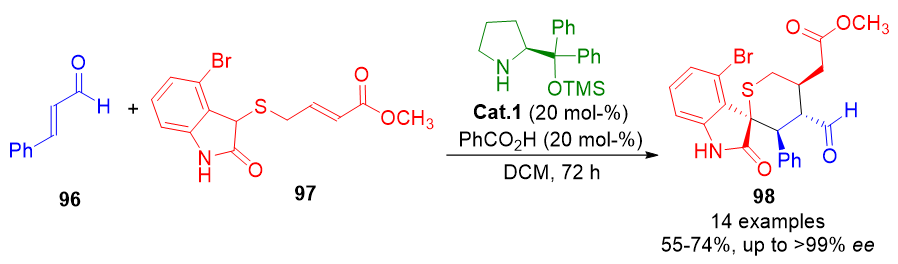
Scheme 21. Sequential iminium–enamine aminocatalytic cascade enabling the enantioselective synthesis of spiro-tetrahydrothiopyran-oxindole scaffolds.
More intricate designs integrate other aminocatalytic pathways. For instance, dual iminium-mediated steps have been executed with di-nucleophilic substrates 99 reacting with dienals 93 in a 1,6- followed by 1,4-addition sequence, catalyzed by Cat.12, resulting in polycyclic products (Scheme 22a).[97] Similarly, a dienamine–iminium cascade has been employed to construct bicyclopyrazolidine frameworks 103 by engaging 1,2-diaza-1,3-dienes with β,β-disubstituted enals 101, combining C–C and C–N bond-forming events under stereocontrol (Scheme 22b).[98]
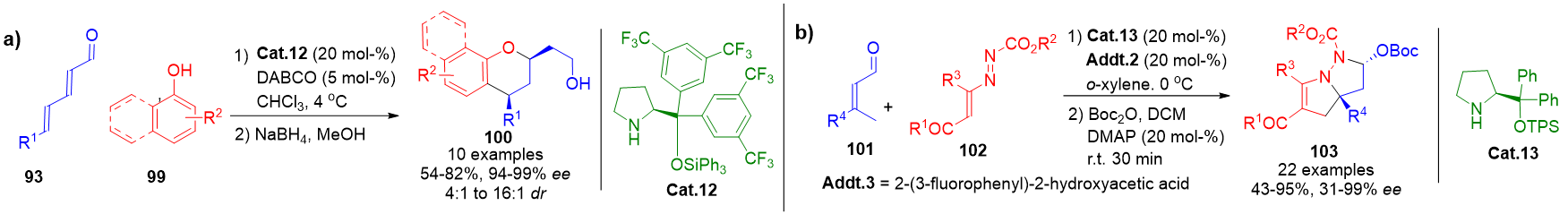
Scheme 22. a) Stereocontrolled 1,6-Friedel–Crafts and 1,4-oxa-Michael cascade via dual iminium activation. b) Construction of bicyclopyrazolidine motifs via a dienamine–iminium sequential activation strategy.
Multicomponent aminocatalytic cascades further expand structural complexity by integrating three or more reactive partners. Carefully selected substrates engage in a series of orchestrated transformations involving alternating activation modes. In one such example, a saturated aldehyde initiates an enamine-catalyzed Michael addition to a Michael acceptor. The resulting intermediate then undergoes iminium-catalyzed conjugate addition with an enal, followed by enamine-mediated ring annulation. These cascades yield diverse scaffolds such as spiropyrazolones 106 and chromans 108, depending on the nature of the Michael acceptor employed (Scheme 23).[99][100]
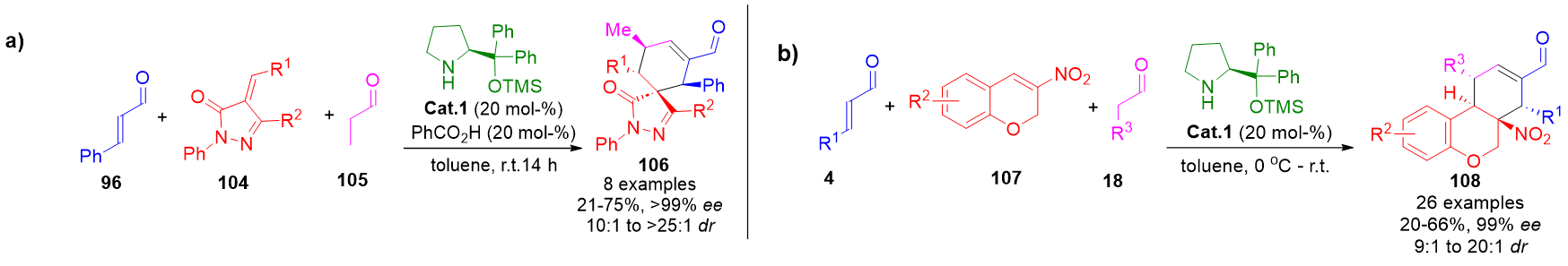
Scheme 23. a) Triple cascade sequence for the formation of chiral spiropyrazolones using alternating enamine and iminium pathways. b) Modular domino reaction generating chromans through an enamine–iminium–enamine cascade using three distinct reactants.
Even greater molecular complexity is accessible through quadruple aminocatalytic cascades, wherein four sequential transformations are executed in a single operation. A representative example includes azabicyclic indole derivatives 110 and tetraaryl-substituted 2-azabicyclo[3.3.0]octadienones 112, synthesized from two equivalents of enals 4 and multifunctional partners 109 or 111, respectively. These sequences proceed via alternating iminium and enamine activation steps, culminating in the formation of intricate polycyclic frameworks (Scheme 24).[101][102]
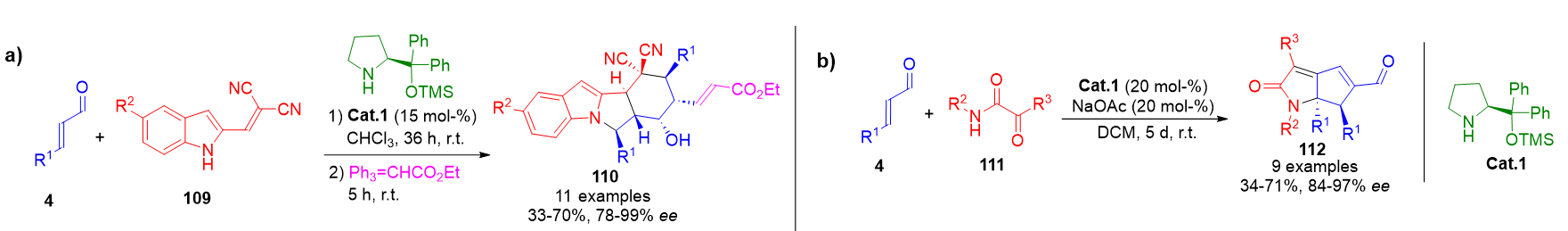
Scheme 24. a) Quadruple cascade comprising aza-Michael, Michael, Michael, and aldol steps leading to complex polycyclic indole derivatives. b) One-pot assembly of tetraaryl 2-azabicyclo[3.3.0]octadienones through iminium–enamine–iminium–enamine activation.
Aminocatalysis, through its multiple activation modes—including enamine, iminium, dienamine, trienamine, tetraenamine, and vinylogous iminium ion catalysis—has become a central strategy in the asymmetric synthesis of privileged molecular frameworks. The ApDOS (Aminocatalytic privileged Diversity-Oriented Synthesis) concept unifies these pathways under a common design principle: the transformation of simple, readily available substrates into structurally diverse and biologically relevant compounds via shared reactive intermediates. Over the past decade, numerous methodological advances and cascade strategies have demonstrated the potential of ApDOS to explore uncharted regions of chemical space. As the field continues to evolve, ApDOS is anticipated to play a growing role in the construction of molecular libraries tailored for biological screening and medicinal chemistry applications, offering a robust and modular approach to molecular innovation.
References
- Cornelius J. O' Connor; Luca Laraia; David R. Spring; Chemical genetics. Chem. Soc. Rev.. 2011, 40, 4332-4345.
- Derek S Tan; Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat. Chem. Biol.. 2005, 1, 74-84.
- Elena Lenci; Antonio Guarna; Andrea Trabocchi; Diversity-Oriented Synthesis as a Tool for Chemical Genetics. Mol.. 2014, 19, 16506-16528.
- Brent R. Stockwell; Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet.. 2000, 1, 116-125.
- Wolfgang H. B. Sauer; Matthias K. Schwarz; Molecular Shape Diversity of Combinatorial Libraries: A Prerequisite for Broad Bioactivity. J. Chem. Inf. Comput. Sci.. 2003, 43, 987-1003.
- Warren Rjd Galloway; David R Spring; Is synthesis the main hurdle for the generation of diversity in compound libraries for screening?. Expert Opin. Drug Discov.. 2009, 4, 467-472.
- Wei Zheng; Natasha Thorne; John C. McKew; Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today. 2013, 18, 1067-1073.
- Bridget K. Wagner; The resurgence of phenotypic screening in drug discovery and development. Expert Opin. Drug Discov.. 2015, 11, 121-125.
- Jesse W.-H. Li; John C. Vederas; Drug Discovery and Natural Products: End of an Era or an Endless Frontier?. Sci.. 2009, 325, 161-165.
- Christopher Cordier; Daniel Morton; Sarah Murrison; Adam Nelson; Catherine O'Leary-Steele; Natural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries. Nat. Prod. Rep.. 2008, 25, 719-37.
- A Harvey; Natural products in drug discovery. Drug Discov. Today. 2008, 13, 894-901.
- Peter Weyerstahl; The Logic of Chemical Synthesis. Flavour Fragr. J.. 1996, 11, 261-262.
- Stuart L. Schreiber; Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Sci.. 2000, 287, 1964-1969.
- Martin D. Burke; Stuart L. Schreiber; A Planning Strategy for Diversity‐Oriented Synthesis. Angew. Chem. Int. Ed. Engl.. 2003, 43, 46-58.
- Richard J. Spandl; Mónica Díaz‐Gavilán; Kieron M. G. O'Connell; Gemma L. Thomas; David R. Spring; Diversity‐oriented synthesis. Chem. Rec.. 2008, 8, 129-142.
- Richard J. Spandl; Andreas Bender; David R. Spring; Diversity-oriented synthesis; a spectrum of approaches and results. Org. Biomol. Chem.. 2008, 6, 1149-1158.
- Warren R.J.D. Galloway; Albert Isidro-Llobet; David R. Spring; Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun.. 2010, 1, 80.
- Cornelius J. O' Connor; Henning S. G. Beckmann; David R. Spring; Diversity-oriented synthesis: producing chemical tools for dissecting biology. Chem. Soc. Rev.. 2012, 41, 4444-4456.
- Sambasivarao Kotha; Arjun S. Chavan; Deepti Goyal; Diversity-Oriented Approaches to Polycyclics and Bioinspired Molecules via the Diels–Alder Strategy: Green Chemistry, Synthetic Economy, and Beyond. ACS Comb. Sci.. 2015, 17, 253-302.
- Miguel Garcia‐Castro; Stefan Zimmermann; Muthukumar G. Sankar; Kamal Kumar; Scaffold Diversity Synthesis and Its Application in Probe and Drug Discovery. Angew. Chem. Int. Ed. Engl.. 2016, 55, 7586-7605.
- Satheeshkumar Reddy Kandimalla; Gowravaram Sabitha; Diversity‐Oriented Synthesis of Oxacyclic Spirooxindole Derivatives through Ring‐Closing Enyne Metathesis and Intramolecular Pauson–Khand (2+2+1) Cyclization of Oxindole Enynes. Adv. Synth. Catal.. 2017, 359, 3444-3453.
- Ohyun Kwon; Seung Bum Park; Stuart L. Schreiber; Skeletal Diversity via a Branched Pathway: Efficient Synthesis of 29 400 Discrete, Polycyclic Compounds and Their Arraying into Stock Solutions. J. Am. Chem. Soc.. 2002, 124, 13402-13404.
- Samantha Caputo; Luca Banfi; Andrea Basso; Andrea Galatini; Lisa Moni; Renata Riva; Chiara Lambruschini; Diversity‐Oriented Synthesis of Various Enantiopure Heterocycles by Coupling Organocatalysis with Multicomponent Reactions. Eur. J. Org. Chem.. 2017, 2017, 6619-6628.
- Sangmi Oh; Seung Bum Park; A design strategy for drug-like polyheterocycles with privileged substructures for discovery of specific small-molecule modulators. Chem. Commun.. 2011, 47, 12754-12761.
- Jonghoon Kim; Heejun Kim; Seung Bum Park; Privileged Structures: Efficient Chemical “Navigators” toward Unexplored Biologically Relevant Chemical Spaces. J. Am. Chem. Soc.. 2014, 136, 14629-14638.
- B. E. Evans; K. E. Rittle; M. G. Bock; R. M. DiPardo; R. M. Freidinger; W. L. Whitter; G. F. Lundell; D. F. Veber; P. S. Anderson; R. S. L. Chang; V. J. Lotti; D. J. Cerino; T. B. Chen; P. J. Kling; K. A. Kunkel; J. P. Springer; J. Hirshfield; Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem.. 1988, 31, 2235-2246.
- David W. C. MacMillan; The advent and development of organocatalysis. Nat.. 2008, 455, 304-308.
- Wendy S. Jen; John J. M. Wiener; David W. C. MacMillan; New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1,3-Dipolar Cycloaddition. J. Am. Chem. Soc.. 2000, 122, 9874-9875.
- Benjamin List; Richard A. Lerner; Carlos F. Barbas; Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc.. 2000, 122, 2395-2396.
- Nobel Prize in Chemistry 2021. The Nobel Prize. Retrieved 2025-4-8
- Peter I. Dalko. Comprehensive Enantioselective Organocatalysis; Peter I. Dalko, Eds.; Wiley: Hoboken, NJ, United States, 2013; pp. -.
- Tushar Janardan Pawar; Hao Jiang; Miguel Ángel Vázquez; Clarisa Villegas Gómez; David Cruz Cruz; Aminocatalytic Privileged Diversity‐Oriented Synthesis (ApDOS): An Efficient Strategy to Populate Relevant Chemical Spaces. Eur. J. Org. Chem.. 2018, 2018, 1835-1851.
- Tushar Janardan Pawar; Suhas Balasaheb Mitkari; Eduardo Peña‐Cabrera; Clarisa Villegas Gómez; David Cruz Cruz; Polyenals and Polyenones in Aminocatalysis: A Decade Building Complex Frameworks from Simple Blocks. Eur. J. Org. Chem.. 2020, 2020, 6044-6061.
- Qi-Yun Huang; Min Shi; Recent advancements in Ni/photoredox dual catalysis for Csp3–Csp3 cross-coupling reactions. Org. Chem. Front.. 2024, 11, 4913-4925.
- Zhenzhi Tan; Qi Yang; Sanzhong Luo; AI molecular catalysis: where are we now?. Org. Chem. Front.. 2025, -, -.
- Antonio Del Vecchio; Arianna Sinibaldi; Valeria Nori; Giuliana Giorgianni; Graziano Di Carmine; Fabio Pesciaioli; Synergistic Strategies in Aminocatalysis. Chem. – A Eur. J.. 2022, 28, e202200818.
- Efraím Reyes; Liher Prieto; Uxue Uria; Luisa Carrillo; Jose L. Vicario; Asymmetric Dual Enamine Catalysis/Hydrogen Bonding Activation. Catal.. 2023, 13, 1091.
- Jun-Long Li; Tian-Yu Liu; Ying-Chun Chen; Aminocatalytic Asymmetric Diels–Alder Reactions via HOMO Activation. Accounts Chem. Res.. 2012, 45, 1491-1500.
- Tushar Janardan Pawar; Suhas Balasaheb Mitkari; Eduardo Peña‐Cabrera; Clarisa Villegas Gómez; David Cruz Cruz; Polyenals and Polyenones in Aminocatalysis: A Decade Building Complex Frameworks from Simple Blocks. Eur. J. Org. Chem.. 2020, 2020, 6044-6061.
- Benjamin List; Enamine Catalysis Is a Powerful Strategy for the Catalytic Generation and Use of Carbanion Equivalents. Accounts Chem. Res.. 2004, 37, 548-557.
- Wolfgang Notz; Fujie Tanaka; Carlos F. Barbas; Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Accounts Chem. Res.. 2004, 37, 580-591.
- Anniina Erkkilä; Inkeri Majander; Petri M. Pihko; Iminium Catalysis. Chem. Rev.. 2007, 107, 5416-5470.
- Mareike C. Holland; Jan Benedikt Metternich; Constantin Daniliuc; W. Bernd Schweizer; Ryan Gilmour; Aromatic Interactions in Organocatalyst Design: Augmenting Selectivity Reversal in Iminium Ion Activation. Chem. – A Eur. J.. 2015, 21, 9937-9937.
- Teresa D. Beeson; Anthony Mastracchio; Jun-Bae Hong; Kate Ashton; David W. C. MacMillan; Enantioselective Organocatalysis Using SOMO Activation. Sci.. 2007, 316, 582-585.
- Søren Bertelsen; Martin Nielsen; Karl Anker Jørgensen; Radicals in Asymmetric Organocatalysis. Angew. Chem. Int. Ed. Engl.. 2007, 46, 7356-7359.
- Dhevalapally B. Ramachary; Y. Vijayendar Reddy; Dienamine Catalysis: An Emerging Technology in Organic Synthesis. Eur. J. Org. Chem.. 2011, 2012, 865-887.
- Vanesa Marcos; José Alemán; Old tricks, new dogs: organocatalytic dienamine activation of α,β-unsaturated aldehydes. Chem. Soc. Rev.. 2016, 45, 6812-6832.
- Indresh Kumar; Panduga Ramaraju; Nisar A. Mir; Asymmetric trienamine catalysis: new opportunities in amine catalysis. Org. Biomol. Chem.. 2012, 11, 709-716.
- Kim Søholm Halskov; Tore Kiilerich Johansen; Rebecca L. Davis; Marianne Steurer; Frank Jensen; Karl Anker Jørgensen; Cross-trienamines in Asymmetric Organocatalysis. J. Am. Chem. Soc.. 2012, 134, 12943-12946.
- Claudio Curti; Lucia Battistini; Andrea Sartori; Franca Zanardi; New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems. Chem. Rev.. 2020, 120, 2448-2612.
- Xin-Yue Gao; Ru-Jie Yan; Ben-Xian Xiao; Wei Du; Łukasz Albrecht; Ying-Chun Chen; Asymmetric Formal Vinylogous Iminium Ion Activation for Vinyl-Substituted Heteroaryl and Aryl Aldehydes. Org. Lett.. 2019, 21, 9628-9632.
- Hao Jiang; Łukasz Albrecht; Karl Anker Jørgensen; Aminocatalytic remote functionalization strategies. Chem. Sci.. 2013, 4, 2287-2300.
- Kateri A. Ahrendt; Christopher J. Borths; David W. C. MacMillan; New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc.. 2000, 122, 4243-4244.
- Wei Zhang; Johan Franzén; Diverse Asymmetric Quinolizidine Synthesis: A Stereodivergent One‐Pot Approach. Adv. Synth. Catal.. 2010, 352, 499-518.
- Gustav Dickmeiss; Kim L. Jensen; Dennis Worgull; Patrick T. Franke; Karl Anker Jørgensen; An Asymmetric Organocatalytic One‐Pot Strategy to Octahydroacridines. Angew. Chem. Int. Ed. Engl.. 2011, 50, 1580-1583.
- Rui Zhou; Qinjie Wu; Mingrui Guo; Wei Huang; Xianghong He; Lei Yang; Fu Peng; Gu He; Bo Han; Organocatalytic cascade reaction for the asymmetric synthesis of novel chroman-fused spirooxindoles that potently inhibit cancer cell proliferation. Chem. Commun.. 2015, 51, 13113-13116.
- Wendy S. Jen; John J. M. Wiener; David W. C. MacMillan; New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1,3-Dipolar Cycloaddition. J. Am. Chem. Soc.. 2000, 122, 9874-9875.
- Liher Prieto; Veronica Juste‐Navarro; Uxue Uria; Ignacio Delso; Efraim Reyes; Tomas Tejero; Luisa Carrillo; Pedro Merino; Jose L. Vicario; Regioselectivity Change in the Organocatalytic Enantioselective (3+2) Cycloaddition with Nitrones through Cooperative Hydrogen‐Bonding Catalysis/Iminium Activation. Chem. – A Eur. J.. 2017, 23, 2764-2768.
- Alberto Fraile; Daniele M. Scarpino Schietroma; Anna Albrecht; Rebecca L. Davis; Karl Anker Jørgensen; Asymmetric Synthesis of Hexahydropyrrolo‐isoquinolines by an Organocatalytic Three‐Component Reaction. Chem. – A Eur. J.. 2012, 18, 2773-2776.
- Cristina Izquierdo; Francisco Esteban; Alejandro Parra; Ricardo Alfaro; José Alemán; Alberto Fraile; José Luis García Ruano; Control of the Dual Reactivity (Iminium-Dienamine) of β-Arylmethyl α,β-Unsaturated Aldehydes in Organocatalytic 1,3-Dipolar Cycloadditions with N-Benzoyl C,N-Cyclic Azomethine Imines. J. Org. Chem.. 2014, 79, 10417-10433.
- Z. G. Hajos, D. R. Parrish, DE 2102623, 1971.
- Zoltan G. Hajos; David R. Parrish; Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem.. 1974, 39, 1615-1621.
- U. Eder, G. R. Sauer, R. Wiechert, DE 2014757, 1971
- Ulrich Eder; Gerhard Sauer; Rudolf Wiechert; Neuartige asymmetrische Cyclisierung zu optisch aktiven Steroid‐CD‐Teilstücken. Angew. Chem.. 1971, 83, 492-493.
- Ulrich Eder; Gerhard Sauer; Rudolf Wiechert; New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angew. Chem. Int. Ed. Engl.. 1971, 10, 496-497.
- Ben Bradshaw; Josep Bonjoch; The Wieland-Miescher Ketone: A Journey from Organocatalysis to Natural Product Synthesis. Synlett. 2011, 23, 337-356.
- You Wang; Shaolin Zhu; Dawei Ma; Organocatalytic Approach to Polysubstituted Piperidines and Tetrahydropyrans. Org. Lett.. 2011, 13, 1602-1605.
- Luyi Zhu; Qiliang Chen; Dan Shen; Weihao Zhang; Cong Shen; Xiaofei Zeng; Guofu Zhong; Enantioselective Construction of Spirocyclic Oxindole Derivatives with Multiple Stereocenters via an Organocatalytic Michael/Aldol/Hemiacetalization Cascade Reaction. Org. Lett.. 2016, 18, 2387-2390.
- Kyatham Srinivas; Nishant Singh; Dinabandhu Das; Dipankar Koley; Organocatalytic, Asymmetric Synthesis of Aza-Quaternary Center of Izidine Alkaloids: Synthesis of (−)-Tricyclic Skeleton of Cylindricine. Org. Lett.. 2016, 19, 274-277.
- Jun-Long Li; Kai-Chuan Yang; Yi Li; Qiang Li; Hong-Ping Zhu; Bo Han; Cheng Peng; Yong-Gang Zhi; Xiao-Jun Gou; Asymmetric synthesis of bicyclic dihydropyrans via organocatalytic inverse-electron-demand oxo-Diels–Alder reactions of enolizable aliphatic aldehydes. Chem. Commun.. 2016, 52, 10617-10620.
- Lei Yang; Wei Huang; Xiang‐Hong He; Ming‐Cheng Yang; Xiang Li; Gu He; Cheng Peng; Bo Han; Stereoselective Synthesis of Hydropyrano[3,2‐b]indoles via Organocatalytic Asymmetric Inverse‐Electron‐Demand Oxa‐Diels–Alder Reaction. Adv. Synth. Catal.. 2016, 358, 2970-2975.
- Si-Li Zhou; Jun-Long Li; Lin Dong; Ying-Chun Chen; Organocatalytic Sequential Hetero-Diels–Alder and Friedel–Crafts Reaction: Constructions of Fused Heterocycles with Scaffold Diversity. Org. Lett.. 2011, 13, 5874-5877.
- Ane Orue; Efraím Reyes; Jose L. Vicario; Luisa Carrillo; Uxue Uria; Enantio- and Diastereoselective Synthesis of Substituted Tetrahydro-1H-isochromanes through a Dynamic Kinetic Resolution Proceeding under Dienamine Catalysis. Org. Lett.. 2012, 14, 3740-3743.
- Anna Albrecht; Jan Bojanowski; Decarboxylative Aminocatalytic Cascade for the Synthesis of Dihydroxanthones. Adv. Synth. Catal.. 2017, 359, 2907-2911.
- Kim Søholm Halskov; Bjarke S. Donslund; Sebastian Barfüsser; Karl Anker Jørgensen; Organocatalytic Asymmetric Formation of Steroids. Angew. Chem. Int. Ed. Engl.. 2014, 53, 4137-4141.
- Cecilia Martín‐Santos; Carlos Jarava‐Barrera; Sandra del Pozo; Alejandro Parra; Sergio Díaz‐Tendero; Rubén Mas‐Ballesté; Silvia Cabrera; José Alemán; Highly Enantioselective Construction of Tricyclic Derivatives by the Desymmetrization of Cyclohexadienones. Angew. Chem. Int. Ed. Engl.. 2014, 53, 8184-8189.
- Rasmus Mose; Magnus E. Jensen; Gert Preegel; Karl Anker Jørgensen; Direct Access to Multifunctionalized Norcamphor Scaffolds by Asymmetric Organocatalytic Diels–Alder Reactions. Angew. Chem. Int. Ed. Engl.. 2015, 54, 13630-13634.
- Zhi-Jun Jia; Hao Jiang; Jun-Long Li; Björn Gschwend; Qing-Zhu Li; Xiang Yin; Julie Grouleff; Ying-Chun Chen; Karl Anker Jørgensen; Trienamines in Asymmetric Organocatalysis: Diels−Alder and Tandem Reactions. J. Am. Chem. Soc.. 2011, 133, 5053-5061.
- Alicia Monleón; Florian Glaus; Stefania Vergura; Karl Anker Jørgensen; Organocatalytic Strategy for the Enantioselective Cycloaddition to Trisubstituted Nitroolefins to Create Spirocyclohexene‐Oxetane Scaffolds. Angew. Chem. Int. Ed. Engl.. 2016, 55, 2478-2482.
- Hao Jiang; David Cruz Cruz; Yang Li; Vibeke Henriette Lauridsen; Karl Anker Jørgensen; Asymmetric Organocatalytic Thio-Diels–Alder Reactions via Trienamine Catalysis. J. Am. Chem. Soc.. 2013, 135, 5200-5207.
- Yankai Liu; Manuel Nappi; Elena Arceo; Silvia Vera; Paolo Melchiorre; Asymmetric Catalysis of Diels–Alder Reactions with in Situ Generated Heterocyclic ortho-Quinodimethanes. J. Am. Chem. Soc.. 2011, 133, 15212-15218.
- Yang Li; Fernando Tur; Rune Pagh Nielsen; Hao Jiang; Frank Jensen; Karl Anker Jørgensen; Enantioselective Formal [4+2] Cycloadditions to 3‐Nitroindoles by Trienamine Catalysis: Synthesis of Chiral Dihydrocarbazoles. Angew. Chem.. 2015, 128, 1032-1036.
- David Cruz Cruz; Rasmus Mose; Clarisa Villegas Gómez; Stine V. Torbensen; Martin S. Larsen; Karl Anker Jørgensen; Organocatalytic Cascade Reactions: Towards the Diversification of Hydroisochromenes and Chromenes through Two Different Activation Modes. Chem. – A Eur. J.. 2014, 20, 11331-11335.
- Jing Gu; Ben‐Xian Xiao; Yu‐Rong Chen; Wei Du; Ying‐Chun Chen; Asymmetric Diels–Alder and Cascade Reaction of Quinone Imine Ketals and 2,4‐Dienals: Construction of Chiral Benzo[de]quinolone Derivatives. Adv. Synth. Catal.. 2016, 358, 296-302.
- Xiao‐Feng Xiong; Quan Zhou; Jing Gu; Lin Dong; Tian‐Yu Liu; Ying‐Chun Chen; Trienamine Catalysis with 2,4‐Dienones: Development and Application in Asymmetric Diels–Alder Reactions. Angew. Chem. Int. Ed. Engl.. 2012, 51, 4401-4404.
- Ben‐Xian Xiao; Wei Du; Ying‐Chun Chen; Asymmetric Dearomatizative Diels–Alder Reaction for the Construction of Hydrodibenzo[b,d]furan Frameworks with Tetrasubstituted Stereogenic Centers. Adv. Synth. Catal.. 2017, 359, 1018-1027.
- Andrea Guerrero-Corella; Juan Asenjo-Pascual; Tushar Janardan Pawar; Sergio Díaz-Tendero; Ana Martín-Sómer; Clarisa Villegas Gómez; José L. Belmonte-Vázquez; Diana E. Ramírez-Ornelas; Eduardo Peña-Cabrera; Alberto Fraile; David Cruz Cruz; José Alemán; BODIPY as electron withdrawing group for the activation of double bonds in asymmetric cycloaddition reactions. Chem. Sci.. 2019, 10, 4346-4351.
- Tushar Janardan Pawar; Edson E. Maqueda-Cabrera; Angel Josabad Alonso-Castro; José Luis Olivares-Romero; David Cruz Cruz; Clarisa Villegas Gómez; Enantioselective synthesis of tetrahydrocarbazoles via trienamine catalysis and their anxiolytic-like activity. Bioorganic Med. Chem. Lett.. 2020, 30, 127063.
- Carlos E. Castillo-Espinoza; Tushar Janardan Pawar; Angel Josabad Alonso-Castro; José Luis Olivares-Romero; Miguel A. Vázquez; David Cruz Cruz; Clarisa Villegas Gómez; First enantioselective synthesis of tetrahydrochromeno[2,3-b]carbazolylacetaldehyde via trienamine catalysis and its biological activity. Chem. Heterocycl. Compd.. 2022, 58, 1-5.
- Julian Stiller; Pernille H. Poulsen; David Cruz Cruz; Jorge Dourado; Rebecca L. Davis; Karl Anker Jørgensen; Organocatalytic [4+2] addition reactions via tetraenamine intermediate. Chem. Sci.. 2014, 5, 2052-2056.
- Qing‐Qing Zhou; You‐Cai Xiao; Xin Yuan; Ying‐Chun Chen; Asymmetric Diels–Alder Reactions of 2,4,6‐Trienals via Tetraenamine Catalysis. Asian J. Org. Chem.. 2014, 3, 545-549.
- Xiao-Long He; Hui-Ru Zhao; Chuan-Qi Duan; Wei Du; Ying-Chun Chen; Remote Asymmetric Oxa-Diels–Alder Reaction of 5-Allylic Furfurals via Dearomatizative Tetraenamine Catalysis. Org. Lett.. 2018, 20, 804-807.
- Xu Tian; Paolo Melchiorre; Control of Remote Stereochemistry in the Synthesis of Spirocyclic Oxindoles: Vinylogous Organocascade Catalysis. Angew. Chem. Int. Ed. Engl.. 2013, 52, 5360-5363.
- Mattia Silvi; Indranil Chatterjee; Yiankai Liu; Paolo Melchiorre; Controlling the Molecular Topology of Vinylogous Iminium Ions by Logical Substrate Design: Highly Regio‐ and Stereoselective Aminocatalytic 1,6‐Addition to Linear 2,4‐Dienals. Angew. Chem. Int. Ed. Engl.. 2013, 52, 10780-10783.
- Pernille H. Poulsen; Stefania Vergura; Alicia Monleón; Danny Kaare Bech Jørgensen; Karl Anker Jørgensen; Controlling Asymmetric Remote and Cascade 1,3-Dipolar Cycloaddition Reactions by Organocatalysis. J. Am. Chem. Soc.. 2016, 138, 6412-6415.
- Shengzheng Wang; Yan Jiang; Shanchao Wu; Guoqiang Dong; Zhenyuan Miao; Wannian Zhang; Chunquan Sheng; Meeting Organocatalysis with Drug Discovery: Asymmetric Synthesis of 3,3′-Spirooxindoles Fused with Tetrahydrothiopyrans as Novel p53-MDM2 Inhibitors. Org. Lett.. 2016, 18, 1028-1031.
- Pernille H. Poulsen; Karla Santos Feu; Bruno Matos Paz; Frank Jensen; Karl Anker Jørgensen; Organocatalytic Asymmetric 1,6‐Addition/1,4‐Addition Sequence to 2,4‐Dienals for the Synthesis of Chiral Chromans. Angew. Chem. Int. Ed. Engl.. 2015, 54, 8203-8207.
- Guang-Yao Ran; Ming Gong; Jing-Fei Yue; Xing-Xing Yang; Su-Lan Zhou; Wei Du; Ying-Chun Chen; Asymmetric Cascade Assembly of 1,2-Diaza-1,3-dienes and α,β-Unsaturated Aldehydes via Dienamine Activation. Org. Lett.. 2017, 19, 1874-1877.
- Alex Zea; Andrea-Nekane R. Alba; Andrea Mazzanti; Albert Moyano; Ramon Rios; Highly enantioselective cascade synthesis of spiropyrazolones. Org. Biomol. Chem.. 2011, 9, 6519-6523.
- Mukesh Kumar; Pankaj Chauhan; Arto Valkonen; Kari Rissanen; Dieter Enders; Asymmetric Synthesis of Functionalized Tricyclic Chromanes via an Organocatalytic Triple Domino Reaction. Org. Lett.. 2017, 19, 3025-3028.
- Dieter Enders; Andreas Greb; Kristina Deckers; Philipp Selig; Carina Merkens; Quadruple Domino Organocatalysis: An Asymmetric Aza‐Michael/Michael/Michael/Aldol Reaction Sequence Leading to Tetracyclic Indole Structures with Six Stereocenters. Chem. – A Eur. J.. 2012, 18, 10226-10229.
- Dieter Enders; Céline Joie; Kristina Deckers; Gerhard Raabe; An Asymmetric Organocatalytic Quadruple Cascade to Tetraaryl-Substituted 2-Azabicyclo[3.3.0]octadienones. Synth.. 2014, 46, 1539-1546.

