 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margarita Martin | + 2450 word(s) | 2450 | 2020-04-14 04:24:14 | | | |
| 2 | Nicole Yin | -11 word(s) | 2439 | 2020-10-28 03:28:26 | | |
Video Upload Options
Long-tailed unconventional class I myosin , Myosin 1E (MYO 1E) and Myosin 1F (MYO1F) are motor proteins that use chemical energy from the hydrolysis of adenosine triphosphate (ATP) to produce mechanical work along the actin cytoskeleton. On the basis of their motor properties and structural features, myosins perform a variety of essential roles in physiological processes such as endocytosis, exocytosis, cell adhesion, and migration. The long tailed unconventional class I myosins are characterized by having a conserved motor head domain, which binds actin and hydrolyzes ATP, followed by a short neck with an isoleucine-glutamine (IQ) motif, which binds calmodulin and is sensitive to calcium, and a tail that contains a pleckstrin homology domain (PH), a tail homology 1 domain (TH1), wherein these domains allow membrane binding, a tail homology 2 domain (TH2), an ATP-insensitive actin-binding site domain, and a single Src homology 3 domain (SH3) susceptible to binding proline rich regions in other proteins. Therefore, these motor proteins are able to bind actin, plasma membrane, and other molecules (adaptor, kinases, membrane proteins) that contribute to their function, ranging from increasing membrane tension to molecular trafficking and cellular adhesion. MYO1E and MYO1F function in host self-defense, with a better defined role in innate immunity in cell migration and phagocytosis.
1. Introduction
MYO1E and MYO1F are long-tailed class I myosins that have similar structures and patterns of tissue expression. Although their similarities reveal some convergences between the two myosins, they have important differences that result in discrepancies in cellular function and their role in diseases. Both MYO1E and MYO1F are expressed mainly in immune system cells. MYO1E has a wider expression pattern than MYO1F, and is highly present in the spleen, mesenteric lymph nodes, and lung, as well as to a lesser extent in the intestines and skin. By contrast, MYO1F is mostly expressed in the spleen, mesenteric lymph nodes, thymus, and lungs[1]. Lymphoid tissues, natural killer cells, macrophages, and dendritic cells express considerable levels of both MYO1E and MYO1F, with selective expression reported in B cells and neutrophils, respectively[1]. Lately, Myo1f has also been reported as being expressed in mast cells[2].
2. MYO1E and MYO1F in Neutrophils
Neutrophils play an important role in innate immunity, and their extravasation in response to an infection or injury is the factor that contributes fastest to the elimination of a pathogen and subsequent wound healing[3][4]. Neutrophil recruitment to the site of inflammation must be carefully regulated because deficient or excessive levels can have severe pathological consequences. As reported recently, neutrophils can contribute to tissue injury by amplifying the inflammatory response and direct release of toxic effectors and assist in the development of many noninfectious diseases, such as lung injury, autoimmune diseases, and cancer[5]. Neutrophil extravasation implies, among other things, proper adhesion to the vascular endothelium and migration to the infected tissue, which depends on actin remodeling and the regulated action of myosins[6][7]. Motile neutrophils exhibit a polarized morphology characterized by the formation of leading edge pseudopods and a highly contractile cell rear known as the uropod[4].
Although MYO1E is barely expressed in neutrophils, it has been shown to be required for their efficient extravasation[8]. Recently, MYO1E-deficient neutrophils were shown to have diminished arrest, spreading, uropod formation, and chemotaxis due to defective actin polymerization and integrin activation. Indeed, β2 integrin-mediated rolling and adhesive interactions are affected in MYO1E knock out neutrophils. This phenotype resulted in increased rolling velocity, decreased firm adhesion, aberrant crawling, and strongly reduced transmigration. Thus, MYO1E appears to regulate the adhesive interactions of neutrophils with the vascular endothelium needed for neutrophil extravasation, reducing both 2D and 3D migration[8].
MYO1F is highly expressed in neutrophils, where it plays an essential role in their migration. Neutrophils from MYO1F-deficient mice showed stronger adhesion to integrin ligands, including intercellular adhesion molecule-1 and fibronectin, and most of this adhesion was mediated by β2 integrin. Indeed, MYO1F-deficient neutrophils exhibited high levels of cell-surface β2 integrin[1]. Given that regulated integrin-mediated adhesion to the vascular endothelium is critical to neutrophil migration to infected tissue, MYO1F-deficient mice unsurprisingly presented higher mortality when exposed to infection by Listeria monocytogenes. Similarly, effects on defective neutrophil migration have been found in SH3-binding protein 2 (3BP2)-knockout mice, which also resulted in a higher mortality to Listeria infection[9]. Interestingly, the adaptor protein 3BP2 has been reported to be a ligand of MYO1F[2]. However, the mechanistic details of how these binding partners regulate neutrophil migration remains to be elucidated.
In the analysis of neutrophil migration in 3D experiments, transmigration and migration in collagen networks showed that neutrophil extravasation into the tissue was also severely compromised in MYO1F-deficient mice due to a defective dynamic deformation of the nucleus[10]. For successful cell migration in these contexts, the nucleus must undergo defined changes in position and shape that are dependent on cytoskeletal dynamics and the mechanical linkage between actin filaments and the nuclear membrane. MYO1F was found to be enriched at the rear and the front ends of the elongated nucleus during the initiation and deformation phases, and it was probably involved in pushing and/or pulling the nucleus through the constriction sites, transmitting force from the cytoskeleton to the inside of the nucleus[10]. Together, these results support the contention that MYO1F is key to host defenses by facilitating neutrophil migration to the site of inflammation.
The impaired neutrophil migration observed in MYO1E- and MYO1F-deficient mice have distinct molecular foundations. In the case of MYO1F, its absence did not lead to reduced neutrophil rolling or adhesion on endothelial cells, a phenomenon that was described in MYO1E-deficient neutrophils[8]. myo1f-mediated neutrophil migration has been reported to be critical to acute neuroinflammation in ischemic stroke, directly affecting outcomes. During the acute phase of a stroke, neutrophils from the peripheral blood are the first to arrive in the ischemic brain, which then attracts other immune cells that exacerbate neuroinflammation in the ischemic tissue[11]. Although further research on dissecting the ligand partners and mechanisms will be important to unraveling the causes of the functional differences between MYO1E and MYO1F, data currently points to long-tailed class I myosins having a key role in neutrophil function.
3. MYO1E and MYO1F in Macrophages
Phagocytosis of invading pathogens and/or cellular debris are processes carried out mainly by macrophages in the different tissues. These events needed for host defense, tissue remodeling, and repair require significant changes in phagocyte morphology that accounts for the coordinated participation of a plethora of molecules involved in adhesion, membrane arrangements, and actin cytoskeleton dynamics[12].
The sensing of infectious danger by macrophages through the ligation of toll-like receptors (TLR) triggers fast and robust cytoskeletal changes, including an integrin-mediated spreading response that is dependent on actin polymerization[13][14]. MYO1E, along with its closely related family member MYO1F, are strongly serine phosphorylated in the tail domain after triggering TLR4, with several sites located in the TH2 domain and one threonine in the PH domain within the TH1 region[15]. Although these data indicate a regulatory mechanism in the action of these myosins in macrophage function against pathogens, no further evidence has been reported. The function of these two myosins seems to be redundant in contributing to lipopolysaccharide-triggered macrophage spreading[16]. In the context of macrophages as antigen presenting cells, MYO1E may control the exocytosis of cytoplasmic vesicles to the plasma membrane (containing major histocompatibility complex class II) through the interaction with the ARF7 effector protein (ARF7EP; also known as ARL14) and contributing to antigen presentation[17]. Consequently, the lack of MYO1E correlates with a deficient antigen-specific T cell proliferation[16].
More recently, it has been reported that MYO1F is induced in colonic macrophages and positively influences αVβ3-integrin accumulation[18]. This process enhances intercellular adhesion between macrophages and stimulates a proinflammatory (M1) phenotype by inducing integrin-linked kinase (ILK)/Protein Kinase B (AKT)/ (mammalian Target of Rapamycin (mTOR) signaling, which, in turn, induces Signal transducers and activators of transcription(STATS), STAT1 and STAT3 activation. Consequently, macrophages lacking MYO1F show reduced intercellular association via integrin-β3 and do not commit to the M1 phenotype. Furthermore, MYO1F upregulation leads to enhanced secretion and production of interleukin-1β and, accordingly, lack of MYO1F has been shown to result in reduced inflammation in a colitis model[18].
More recent data have shown that MYO1E and MYO1F are both required for efficient Fc receptors (FcR)-mediated phagocytosis[19]. Engagement of FcR on the phagocyte with antibodies on the target surface induces phagocytic cup formation (an actin-rich cup-like structure) to engulf the target. The plasma membrane is extended around the phagocytic cup and the ensuing closure of the cup results in phagosome formation where further processing of the target will occur. All these steps require active cytoskeleton dynamics and mechanical forces[12][20]. Macrophages form circular dynamic actin waves in the extending arms of the phagocytic cup, an event linked to phagocytosis[21]. MYO1E and F are reported as being recruited to the phagocytic cup, and they are found to be enriched in the punctate actin that makes up the wave. This location is dependent on the actin-binding via the motor domain, as well as the TH2 domain (via phospholipid membrane binding through the basic amino acids present in this domain), and thus it is exclusive to long-tailed unconventional class I myosins. The study of Barger et al. suggests a model where MYO1E and F function to tether the plasma membrane to the actin to successfully anchor the target to the cell, allowing actin polymerization within the cup to progress and finish internalization. The absence of MYO1E-F alters the local membrane tension and actin polymerization, resulting in an actin-dense phagocytic cup that leads to a slower closure of the phagocytic cup and lower rate of phagocytosis[19].
4. MYO1F in Mast Cells
Mast cells are essential effector cells in both the innate and adaptive arms of the immune system. They rely on their ability to migrate to inflammatory sites and release specific mediators stored in preformed granules or synthesized de novo if they are to function[22]. These processes are highly regulated by signaling events and precise cytoskeletal dynamics. Mast cells are resident in tissues, meaning that their migration as progenitors is needed before recruitment to inflammatory target tissue is possible[23]. Increased numbers of mast cells in inflamed tissue occur not only in bacterial and parasitic infection, but also in asthma and urticarial[24]. In these settings, mast cells recognize chemotactic stimuli and trigger signaling cascades that lead to integrin activation, adhesion, and migration. Stem cell factor (the tyrosine-protein kinase KIT receptor ligand) is the key chemotactic factor in mast cell proliferation, survival, homing, and migration[25]. Recently, our group showed that MYO1F is expressed in mast cells, where it colocalizes with the cortical actin ring[2]. We also found that 3BP2 interacts with Myo1f. The cytoplasmic adaptor protein 3BP2 contains a PH domain, SH3-binding proline-rich regions, and a C-terminal SH2 domain[26], and it has roles in mast cell degranulation[27], survival[28], and migration[2]. Interestingly, the 3BP2–MYO1F interaction is modulated by KIT receptor signaling, possibly by the increase of phosphoinositide 3-kinase (PI3K) activity and consequently the production of phosphatidylinositol-3,4,5-triphosphate, ligands of the PH domains contained in 3BP2 and MYO1F, resulting in a major recruitment of both at the plasma membrane.
Consequently, KIT inhibition alters MYO1F and 3BP2 translocation to the membrane, and subsequently colocalization of both molecules[2]. In the context of KIT signaling, the absence of Myo1f or 3BP2 impairs integrin-mediated mast cell adhesion and migration. Myo1f silencing specifically achieves this by decreasing the expression of two integrin β chains on the cell surface, β1 (Cluster of Differentiation 29) and β7, usually coupled with the α4 chain[2].
These data point to a model where MYO1F could serve as a link between the actin cytoskeleton and the localization and function of integrin in the cell membrane after the activation of KIT receptors (Figure 1). We further hypothesize that MYO1F could modulate integrin by regulating the cortical actin mesh, on the basis of evidence that the cortical actin ring is necessary for mast cell migration[29]. Myo1E has not been reported in mast cells to date.
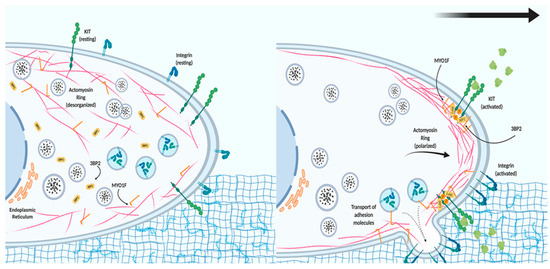
Figure 1. The role of MYO1F in mast cell migration. In resting conditions, mast cells express tyrosine-protein kinase KIT receptor and different integrins on the cell membrane. The actin cytoskeleton and MYO1F are distributed along the cell membrane and in the cytoplasm, whereas Src homology 3 domain (SH3)-binding protein 2 (3BP2) is mostly found in the cytoplasm. Activation of the KIT receptor initiates actin remodeling, which generates the leading edge (consisting of actin filaments) necessary for cell movement. MYO1F is necessary for the secretion and localization of activated integrin molecules that will induce adhesion to the extracellular matrix and aid cell migration.
5. MYO1E in B Lymphocytes
The migration of lymphocytes to lymph nodes is a crucial step for the immune response to encounter antigens[30]. The adhesion of lymphocytes to high endothelial venules and their migration through that network are regulated by adhesins, integrins, and chemokines, as well as the actin cytoskeleton[31][32]. In lymphocytes, the expression of long-tailed unconventional class I myosins appears to be restricted to MYO1E. Its expression is especially high in B lymphocytes, but until recently, its role has been elusive. Consistent with its role in other immune cells, Myo1e has been reported as being critical for the recruitment and adhesion of activated B cells to the inguinal lymph node. Rolling and cellular transmigration are affected in activated B lymphocytes from MYO1E KO mice by a reduction of cell spreading (a mechanism used to maximize cellular contact to allow transmigration) due to a lack of CARMIL (capping protein, Arp2/3, and Myosin-I linker), which is important in cell migration and a ligand of MYO1E[33][34]. On the other hand, activated B cells from MYO1E KO mice have reduced levels of LFA-1 (Lymphocyte Function associated Antigen 1), CD44, and VLA-4 (Very Late Antigen-4) at the plasma membrane, suggesting that MYO1E is playing a role in vesicle trafficking of these adhesion molecules. The molecular mechanism involves the focal adhesion kinase (FAK), which has a role in integrin-mediated signal transduction[35]. FAK activity is reduced in activated B lymphocytes from MYO1E KO mice as well as AKT and the RAC-1 GTPase, both dependent on PI3K activity[34]. An explanation for the reduction in FAK activity in the MYO1E KO model may be by the fact that MYO1E interacts with FAK and promotes its autophosphorylation upon stimulation with CXCL12 in activated B cells[34].
References
- Sangwon V. Kim; Wajahat Z. Mehal; Xuemei Dong; Volkmar Heinrich; Marc Pypaert; Ira Mellman; Micah Dembo; Mark S. Mooseker; Dianqing Wu; Richard A. Flavell; et al. Modulation of Cell Adhesion and Motility in the Immune System by Myo1f. Science 2006, 314, 136-139, 10.1126/science.1131920.
- Arnau Navinés-Ferrer; Erola Ainsua-Enrich; Eva Serrano-Candelas; Joan Sayós; M. Martín; Myo1f, an Unconventional Long-Tailed Myosin, Is a New Partner for the Adaptor 3BP2 Involved in Mast Cell Migration. Frontiers in Immunology 2019, 10, null, 10.3389/fimmu.2019.01058.
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707.
- Hind, L.E.; Vincent, W.J.B.; Huttenlocher, A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev. Cell 2016, 38, 161–169.
- Selina K Jorch; Selina K Jorch Paul Kubes; An emerging role for neutrophil extracellular traps in noninfectious disease. Nature Medicine 2017, 23, 279-287, 10.1038/nm.4294.
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704.
- Schnoor, M.; García Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997.
- Eduardo Vadillo; Sandra Chánez-Paredes; Hilda Vargas-Robles; Idaira María Guerrero-Fonseca; Ramón Castellanos-Martínez; Alexander García-Ponce; Porfirio Nava; Daniel Alberto Girón-Pérez; Leopoldo Santos-Argumedo; Michael Schnoor; et al. Intermittent rolling is a defect of the extravasation cascade caused by Myosin1e-deficiency in neutrophils. Proceedings of the National Academy of Sciences 2019, 116, 26752-26758, 10.1073/pnas.1902502116.
- Grace Chen; Ioannis Dimitriou; Laura Milne; Karl S. Lang; Philipp A. Lang; Noah Fine; Pamela S. Ohashi; Paul Kubes; Robert Rottapel; The 3BP2 Adapter Protein Is Required for Chemoattractant-Mediated Neutrophil Activation. The Journal of Immunology 2012, 189, 2138-2150, 10.4049/jimmunol.1103184.
- Melanie Salvermoser; Robert Pick; Ludwig T. Weckbach; Annette Zehrer; Phillip Löhr; Maik Drechsler; Markus Sperandio; Oliver Soehnlein; Barbara Walzog; Myosin 1f is specifically required for neutrophil migration in 3D environments during acute inflammation. Blood 2018, 131, 1887-1898, 10.1182/blood-2017-10-811851.
- Yan Wang; Haojie Jin; Weifang Wang; Feng Wang; Heng Zhao; Myosin1f-mediated neutrophil migration contributes to acute neuroinflammation and brain injury after stroke in mice. Journal of Neuroinflammation 2019, 16, 1-9, 10.1186/s12974-019-1465-9.
- Spencer A. Freeman; Sergio Grinstein; Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunological Reviews 2014, 262, 193-215, 10.1111/imr.12212.
- Blander, J.M.; Medzhitov, R. Regulation of Phagosome Maturation by Signals from Toll-Like Receptors. Science 2004, 304, 1014–1018.
- Wenzel, J.; Held, C.; Palmisano, R.; Teufel, S.; David, J.-P.; Wittenberg, T.; Lang, R. Measurement of TLR-Induced Macrophage Spreading by Automated Image Analysis: Differential Role of Myd88 and MAPK in Early and Late Responses. Front. Physiol. 2011, 2, 71.
- Gabriele Weintz; Jesper V Olsen; Katja Frühauf; Magdalena Niedzielska; Ido Amit; Jonathan Jantsch; Jörg Mages; Cornelie Frech; Lars Dölken; Matthias Mann; et al.Roland Lang The phosphoproteome of toll‐like receptor‐activated macrophages. Molecular Systems Biology 2010, 6, 371-371, 10.1038/msb.2010.29.
- Jens Wenzel; Jessica L. Ouderkirk; Mira Krendel; Roland Lang; Class I myosinMyo1eregulates TLR4-triggered macrophage spreading, chemokine release, and antigen presentation via MHC class II. European Journal of Immunology 2014, 45, 225-237, 10.1002/eji.201444698.
- Petra Paul; Tineke Van Den Hoorn; Marlieke L.M. Jongsma; Mark J. Bakker; Rutger Hengeveld; Lennert Janssen; Peter Cresswell; David A. Egan; Marieke Van Ham; Anja Ten Brinke; et al.Huib OvaaRoderick L. BeijersbergenCoenraad KuijlJacques Neefjes A Genome-wide Multidimensional RNAi Screen Reveals Pathways Controlling MHC Class II Antigen Presentation. Cell 2011, 145, 268-283, 10.1016/j.cell.2011.03.023.
- Zayda L. Piedra-Quintero; Carolina Serrano; Nicolás Villegas-Sepúlveda; José L. Maravillas-Montero; Sandra Romero-Ramírez; Mineko Shibayama; Oscar Medina-Contreras; Porfirio Nava; Leopoldo Santos-Argumedo; Myosin 1F Regulates M1-Polarization by Stimulating Intercellular Adhesion in Macrophages. Frontiers in Immunology 2019, 9, 3118, 10.3389/fimmu.2018.03118.
- Sarah R. Barger; Nicholas S. Reilly; Maria S. Shutova; Qingsen Li; Paolo Maiuri; John M. Heddleston; Mark S. Mooseker; Richard A. Flavell; Tatyana Svitkina; Patrick W. Oakes; et al.Mira KrendelNils C. Gauthier Membrane-cytoskeletal crosstalk mediated by myosin-I regulates adhesion turnover during phagocytosis. Nature Communications 2019, 10, 1-18, 10.1038/s41467-019-09104-1.
- Erick García-García; Carlos Rosales; Signal transduction during Fc receptor-mediated phagocytosis.. Journal of Leukocyte Biology 2002, 72, 357–362.
- Thomas A. Masters; Michael P. Sheetz; Nils C. Gauthier; F-actin waves, actin cortex disassembly and focal exocytosis driven by actin-phosphoinositide positive feedback. Cytoskeleton 2016, 73, 180-196, 10.1002/cm.21287.
- Stephen J. Galli; Janet Kalesnikoff; Michele A. Grimbaldeston; Adrian M. Piliponsky; Cara M.M. Williams; Mindy Tsai; MAST CELLS AS “TUNABLE” EFFECTOR AND IMMUNOREGULATORY CELLS: Recent Advances. Annual Review of Immunology 2005, 23, 749-786, 10.1146/annurev.immunol.21.120601.141025.
- Joakim S. Dahlin; Jenny Hallgren; Mast cell progenitors: Origin, development and migration to tissues. Molecular Immunology 2015, 63, 9-17, 10.1016/j.molimm.2014.01.018.
- Alasdair M. Gilfillan; Michael A. Beaven; Regulation of Mast Cell Responses in Health and Disease. Critical Reviews™ in Immunology 2011, 31, 475-530, 10.1615/critrevimmunol.v31.i6.30.
- Ivana Halova; Lubica Dráberová; Petr Draber; Mast Cell Chemotaxis – Chemoattractants and Signaling Pathways. Frontiers in Immunology 2012, 3, 119, 10.3389/fimmu.2012.00119.
- R Ren; B J Mayer; P Cicchetti; D Baltimore; Identification of a ten-amino acid proline-rich SH3 binding site.. Science 1993, 259, 5098.
- Erola Ainsua-Enrich; Damiana Álvarez-Errico; Alasdair M. Gilfillan; César Picado; Joan Sayós; Juan Rivera; M. Martín; The adaptor 3BP2 is required for early and late events in FcεRI signaling in human mast cells.. The Journal of Immunology 2012, 189, 2727-2734, 10.4049/jimmunol.1200380.
- Erola Ainsua-Enrich; Eva Serrano-Candelas; Damiana Álvarez-Errico; César Picado; Joan Sayós; Juan Rivera; Margarita Martín; The Adaptor 3BP2 Is Required for KIT Receptor Expression and Human Mast Cell Survival. The Journal of Immunology 2015, 194, 4309-4318, 10.4049/jimmunol.1402887.
- Pavel Dráber; Petr Draber; Membrane-Cytoskeleton Dynamics in the Course of Mast Cell Activation. Advanced Structural Safety Studies 2014, 1220, 219-237, 10.1007/978-1-4939-1568-2_14.
- Luka Mesin; Jonatan Ersching; Gabriel D. Victora; Germinal Center B Cell Dynamics. Immunity 2016, 45, 471-482, 10.1016/j.immuni.2016.09.001.
- Girard, J.-P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773.
- Mionnet, C.; Sanos, S.L.; Mondor, I.; Jorquera, A.; Laugier, J.-P.; Germain, R.N.; Bajénoff, M. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood 2011, 118, 6115–6122.
- Liang, Y.; Niederstrasser, H.; Edwards, M.; Jackson, C.E.; Cooper, J.A. Distinct Roles for CARMIL Isoforms in Cell Migration. Mol. Biol. Cell 2009, 20, 5290–5305.
- Girón-Pérez, D.A.; Vadillo, E.; Schnoor, M.; Santos-Argumedo, L. Myo1e modulates the recruitment of activated B cells to inguinal lymph nodes. J. Cell Sci. 2020, 133, jcs235275.
- J Thomas Parsons; Karen H Martin; Jill K Slack; Joan M Taylor; Scott A Weed; Focal Adhesion Kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606-5613, 10.1038/sj.onc.1203877.

