 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mariana Spetea | -- | 3038 | 2024-04-16 10:00:56 | | | |
| 2 | Mariana Spetea | Meta information modification | 3038 | 2024-04-16 13:10:38 | | |
Video Upload Options
Diverse chemical and pharmacological strategies are currently being explored to minimize the unwanted side effects of currently used opioid analgesics while achieving effective pain relief. The use of multitarget ligands with activity at more than one receptor represents a promising therapeutic approach.
1. Introduction
Pain is an unsolved medical condition, and it is among the most prevalent and debilitating human illnesses [1][2]. Pain is not only a disabling symptom of many medical conditions, but also a disease state in its own right. Central goals in pain control are to provide analgesia of adequate efficacy and to reduce the complications of currently available drugs. Opioid analgesics have been on the frontline of pain management for many years. However, available opioid pain medications cause serious side effects, including a high burden of abuse and addiction [3][4][5]. The rapid increase in the use of opioid drugs in the United States has been termed the “opioid crisis”, with more than 80,000 opioid-related deaths reported in 2021 [6].
The analgesic effects of opioids are mediated through four receptors, namely mu-(MOR), kappa-(KOR), delta-(DOR) and nociceptin/orphanin FQ (NOP) [5][7]. All four opioid receptors are members of the seven-transmembrane-spanning G protein-coupled receptor (GPCR) family and are expressed throughout the central and peripheral nervous systems (CNS and PNS, respectively) and non-neuronal tissues [5][7]. The MOR, which is the main target of clinically used opioid medications, is central for analgesia, but it is also responsible for undesirable side effects, particularly addiction and abuse liabilities [5][7][8].
Because of the inadequate benefit/risk ratio of currently available opioids, together with the ongoing opioid epidemic, there is a huge quest for strategies to discover new opioid analgesics. Alternative chemical and pharmacological approaches are therefore explored to mitigate the deleterious effects of MOR agonists and to limit their abuse and misuse [9][10][11][12][13][14][15][16][17][18][19], amongst which are multifunctional drugs. The concept of ‘one molecule, multiple targets’ has received increased attention in opioid drug discovery as a promising strategy to generate new analgesics with enhanced effectiveness and reduced unwanted side effects. Accumulated preclinical data were reported on multifunctional analgesics targeting opioid/opioid and opioid/non-opioid receptors, including both small molecules and peptide-based ligands [20][21][22][23][24][25][26][27]. Furthermore, the application of two or more biologically active components in the form of a single molecule can significantly improve the physicochemical and/or pharmacokinetic properties of CNS activity.
Whereas opioid analgesics and other addictive substances (e.g., cocaine and cannabinoids) act directly on their specific receptors, they also indirectly influence the dopamine system that controls reward behavior [5][28][29][30]. The reward system consists of dopaminergic neurons, whose activation causes an increase in dopamine release in mesolimbic structures, resulting in a rewarding effect. Among the dopamine receptor subtypes, the D1, D2 and D3 receptors (D1R, D2R and D3R, respectively) have been reported to play important roles in the reinforcement of drug-seeking behavior [31][32][33][34][35][36][37]. Recent data suggest using dopamine D1R or D3R preferring modulators to prevent morphine tolerance and to reduce the duration of morphine withdrawal symptoms [38].
Thus, in light of recent research, bifunctional ligands encompassing pharmacophores targeting both opioid and dopamine receptors received interest as potentially safer analgesics. Designing dual opioid–dopamine receptor ligands may be an attractive way to balance the unwanted side effects derived from their individual components. The first described bifunctional opioid–dopamine receptor ligands were small molecules with a dual-target binding on the MOR and dopamine D3R, with a MOR agonism and D3R antagonism/partial agonism [39][40]. Peptides and peptide analogues offer several advantages over small molecules in various biomedical applications, including pain [41][42][43][44][45]. Some benefits of peptides include high target specificity, enhanced potency, improved selectivity, reduced off-target engagement, reduced toxicity and modularity for future pharmacokinetic optimization and formulation.
The bifunctional peptide-based hybrid LENART01 combines an MOR pharmacophore and a dopamine D2R pharmacophore [46]. The design strategy for LENART01 was based on merging dermorphin and ranatensin by following the principle of pharmacophore fusion (Figure 1).

Figure 1. The amino acid sequence of LENART01. The dermorphin pharmacophore is framed in blue, and the ranatensin analogue pharmacophore is framed in red.
The N-terminal pharmacophore of LENART01 consisted of dermorphin with an amino acid sequence of YdAFGYPS, and the C-terminus was constructed from ranatensin, which was modified, resulting in a sequence of GHFM. The opioid pharmacophore dermorphin is a heptapeptide isolated from the skin of the Amazon frog Phyllomedusa sauvagei [47]. Dermorphin is a selective MOR agonist and potent antinociceptive used as a lead for the design of dermorphin analogues [48][49][50][51]. In early clinical studies, dermorphin administered via the intrathecal (i.t.) route had increased effectiveness than morphine in treating postoperative pain [52]. Ranatensin is an undecapeptide that is first isolated from the skin of the frog Rana pipiens [53][54]. Ranatensin belongs to the bombesin-like peptide family and shares significant homology with bombesin [55][56]. In vitro and in vivo studies described ranatensin to have activity at the dopamine D2R and to attenuate pain behavior in mice after central intracerebroventricular (i.c.v.) administration, with dopamine neurotransmission contributing to the antinociceptive action of ranatensin [57][58].
In vitro binding studies established LENART01 to bind to and activate both MOR and D2R in rat brains and spinal cords. Furthermore, it showed high selectivity to the D2R compared to D1R [46]. LENART01 was also described to have antimicrobial activity against different E. coli strains [59]. With the crystal structures of the active human MOR and available dopamine D2R, an initial in silico evaluation using molecular docking and molecular dynamics (MD) simulations was conducted on the binding mode and interaction mechanisms of LENART01 with the two GPCRs [46].
Because of the interesting profile of LENART01, investigations into its pharmacology were warranted, including (a) in vitro profile to human opioid receptors (binding, selectivity and agonist activities), (b) in vivo behavioral properties in mouse models of acute and inflammatory pain after subcutaneous (s.c.) administration, together with the potential for inducing opioid liabilities of locomotor dysfunction and withdrawal response. And (c) studies on the mechanism underlying the antinociceptive effect of LENART01.
2. In Vitro Pharmacological Profile of LENART01 to the Human Opioid Receptors
The specific binding of LENART01 to the rat MOR and rat dopamine D2R in the brain and spinal cord membrane preparations and its high selectivity to the dopamine D2R compared to D1R was initially determined [46]. Because the human MOR is the ultimate target of therapeutic opioid drugs, LENART01’s binding properties to the human MOR and the other opioid receptors, DOR, KOR and NOP receptors, were determined using in vitro radioligand competition binding to membrane preparations from Chinese hamster ovary (CHO) cells stably expressing the recombinant opioid receptors [60]. The opioid binding profile of LENART01 was compared to that of dermorphin.
LENART01 and dermorphin produced a concentration-dependent inhibition of the selective opioid radioligands, [3H]DAMGO, [3H]DPDPE and [3H]U69,593, binding from the human MOR, DOR and KOR, respectively (Figure 2). Binding studies of the human NOP receptor using [3H]nociceptin indicated no substantial binding of LENAR01 and dermorphin at the concentration of 10 µM (Figure 2D). LENART01 bound to the human MOR with high affinity and reduced binding to the human DOR and KOR (Table 1). In competition binding assays, dermorphin also showed high binding affinity to the MOR and selectivity for the MOR, which aligned with reported data on the neuronal MOR in rat brains [61][62] or to the recombinant MOR expressed in CHO or human embryonic kidney (HEK293) cells [63][64]. When compared to the binding profile of dermorphin, LENART01 displayed a decreased affinity to the human MOR by around six-fold, whereas a maximum of a two-fold decrease was calculated for binding to the human DOR and KOR (Table 1). LENART01 maintained the MOR selectivity of dermorphin, though with lower selectivity ratios for MOR versus the other opioid receptor subtypes.
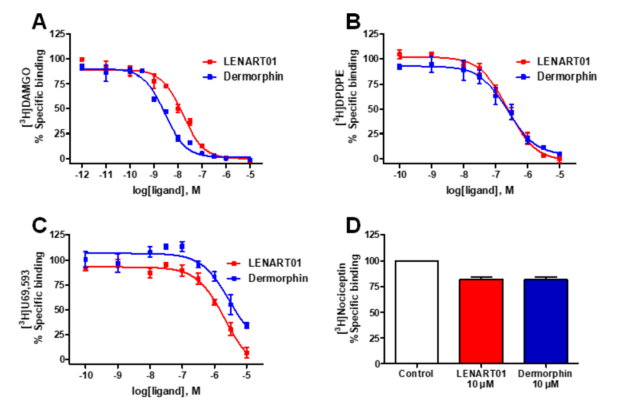
Figure 2. In vitro binding of LENART01 and dermorphin to human opioid receptors determined in radioligand competition binding assays. (A) Concentration-dependent inhibition of [3H]DAMGO binding to CHO-hMOR cell membranes. (B) Concentration-dependent inhibition of [3H]DPDPE binding to CHO-hDOR cell membranes. (C) Concentration-dependent inhibition of [3H]U69,593 binding to CHO-hKOR cell membranes. (D) Specific binding to human NOP receptor using [3H]nociceptin and CHO-hNOP cell membranes. Values represent means ± SEM (n = 3 independent experiments).
Table 1. In vitro binding affinities of LENART01 and dermorphin to human opioid receptors.
|
Ligand |
Opioid Receptor Binding (Ki, nM) |
Selectivity Ki Ratios |
|||||
|
MOR |
DOR |
KOR |
NOP |
DOR/MOR |
KOR/MOR |
NOP/MOR |
|
|
LENART01 |
11.0 ± 1.5 |
227 ± 47 |
1306 ± 226 |
>10,000 |
21 |
119 |
>900 |
|
Dermorphin |
1.89 ± 0.2 |
165 ± 23 |
2710 ± 873 |
>10,000 |
87 |
1434 |
>5000 |
The values were determined in radioligand competition binding assays using membranes from CHO cells stably expressing one of the human opioid receptors, MOR, DOR, KOR or NOP receptor. The values represent the means ± SEM (n = 3 independent experiments).
LENART01 was depicted as an agonist to the rat MOR in the brain and spinal cord preparations using the guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding assay [46]. The agonist activity of LENART01 to the human MOR using the [35S]GTPgS binding assay with CHO-hMOR cell membranes was established and compared to activity of dermorphin. LENART01 increased [35S]GTPγS binding to the human MOR in a concentration-dependent manner, demonstrating full efficacy compared to DAMGO and dermorphin (Figure 3). LENAR01 displayed potent MOR agonist activity, with a potency, ED50 value, only two-fold compared to DAMGO, and a five-fold reduced potency compared to dermorphin (Table 2).
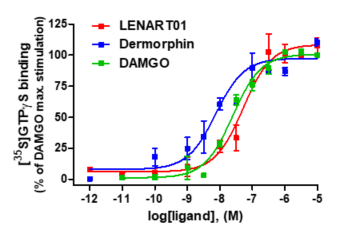
Figure 3. In vitro agonist activity of LENART01 and dermorphin to human MOR. Concentration-dependent stimulation of [35S]GTPγS binding by LENART01, dermorphin and DAMGO determined in the [35S]GTPγS binding assay using membranes from CHO cells expressing the human MOR. Percentage stimulation is presented relative to maximum simulation of reference MOR agonist, DAMGO. Values represent means ± SEM (n = 3–4 independent experiments).
Table 2. In vitro agonist activities of LENART01 and dermorphin to human MOR.
|
Ligand |
EC50 (nM) |
% stim. |
|
LENART01 |
48.5 ± 10.6 |
110 ± 7 |
|
Dermorphin |
9.55 ± 2.58 |
104 ± 8 |
|
DAMGO |
22.2 ± 4.7 |
100 |
Determined in [35S]GTPγS binding assay using membranes from CHO cells stably expressing human MOR. Percentage stimulation (% stim.) is relative to reference MOR agonist DAMGO. Values represent means ± SEM (n = 3–4 independent experiments).
3. In Vivo Pharmacological Profile of LENART01
Antinociceptive Efficacy
Both peptides, dermorphin and ranatensin, the two pharmacophores comprising LENART01, have antinociceptive effects [48][57]. Antinociceptive properties of LENART01 were assessed in a mouse model of acute nociceptive pain using the radiant heat tail-flick test [65] after s.c. administration in mice, where tail withdrawal latencies of mice to thermal stimulation were measured [66]. Subcutaneous administration of LENART01 to mice did not produce a significant increase in the tail-flick latencies at any time point and tested doses (Figure 4A). In the same pain assay, conventional opioids including morphine, oxycodone, oxymorphone, buprenorphine and fentanyl were well-established to induce potent antinociceptive effects following systemic administration to rodents [66][67][68][69]. Activity in the tail-flick test suggests that a drug acts via CNS and may reflect spinal activity [70].
The therapeutic antinociceptive potential of LENART01 was also investigated in a model of inflammatory pain using the formalin test [71]. Pain behavior was assessed in mice that received an s.c. injection of formalin solution to the plantar surface of the right hind paw as described previously [60].
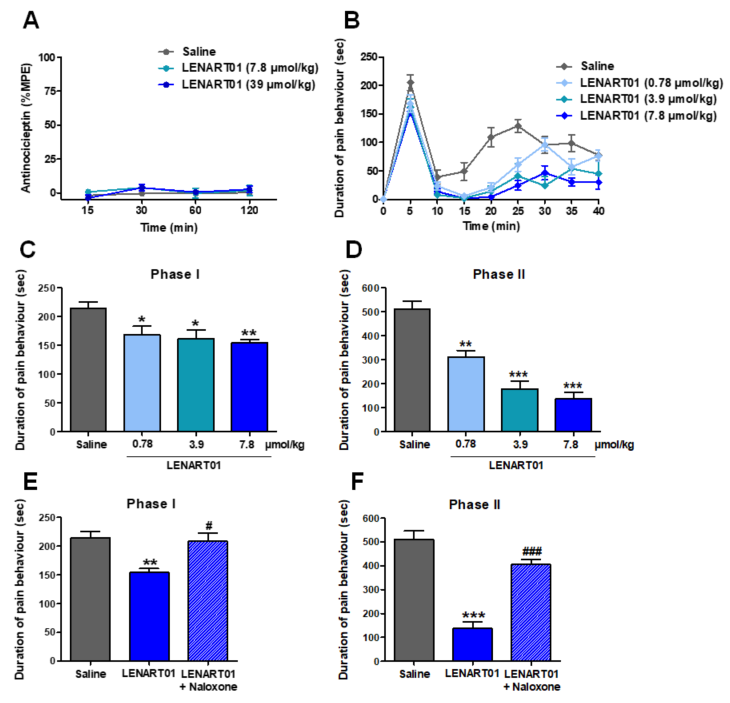
Figure 4. Effect of LENART01 in models of acute nociceptive pain and inflammatory pain after s.c. administration in mice. (A) Radiant heat tail-flick test. Groups of mice received s.c. saline or different doses of LENART01 (3.9 and 7.8 µmol/kg), and tail-flick latencies were measured before and after drug administration at different times. Antinociceptive response is expressed as % of maximum possible effect (%MPE). Values represent means ± SEM (n = 5 mice per group). (B–F) Formalin test. Groups of mice received s.c. saline (control) or different doses of LENART01 (0.78, 3.9 and 7.8 µol/kg) before an intraplantar injection of the formalin solution in right hind paw. Duration of pain behavior was monitored for 40 min, determined as amount of time (in seconds, sec) each animal spent licking, biting, lifting and flinching formalin-injected paw. Time course of pain behavior in formalin test on mice treated with LENART01 (B). Dose-dependent antinociceptive effect of LENART01 during nociceptive Phase I (C) and inflammatory Phase II (D) of the formalin test. (E,F) Opioid antagonism by naloxone on antinociceptive effect of LENART01 during Phase I and Phase II. Mice were s.c. pre-treated with naloxone (1 mg/kg), 15 min before s.c. administration of LENART01 (7.8 µmol/kg). Values represent means ± SEM (n = 6–8 mice per group). *p < 0.05, ** p < 0.01, *** p < 0.001 vs. saline group; # p < 0.01, ### p <0.001 vs. LENART01-treated group; one-way ANOVA with Tukey’s post hoc test.
The systemic s.c. administration of LENAR01 produced time- and dose-dependent reductions in pain behavior in the formalin-injected mice, determined as the amount of time each animal spent licking, biting, lifting and flinching the formalin-injected paw (Figure 4B). LENART01 attenuated the pain behavior during the nociceptive Phase I and inflammatory Phase II of the formalin assay with a significant effect at all tested doses (Figure 4C,D). The calculated antinociceptive effective dose ED50 value of LENART01 in the inflammatory Phase II of the formalin test was 1.60 µmol/kg. The antinociceptive ED50 value of morphine in the formalin test in mice after s.c. administration was reported to be 6.44 µmol/kg [60]. LENART01 shows antinociceptive potency around 4-fold higher than morphine in mice with inflammatory pain. The involvement of the opioid receptors in the antinociceptive effect of LENART01 in the formalin test was investigated using the opioid antagonist naloxone (Figure 4E,F). Pre-treating mice with naloxone resulted in a significant and complete reversal of the antinociceptive response of LENART01 in both Phases I and II, signifying that opioid receptors, specifically the MOR, given the MOR selectivity (Table 1), are involved in LENART01’s in vivo agonist activity.
Motor Coordination
Conventional opioid analgesics, such as morphine, oxycodone, oxymorphone, buprenorphine and fentanyl, are known to produce sedation and alter locomotor activity, which represent undesirable side effects that limit their clinical usefulness [5][66][67][68]. The effect of LENART01 on motor coordination and its potential to induce sedation in mice after s.c. administration were inbestigated in the rotarod test, a well-established model for evaluating the loss of coordinated locomotion [72]. LENART01 did not produce changes in the motor behaviors of mice, as no significant alterations in the rotarod latencies were measured at any time point and tested dose compared to the saline-treated animals. These observations establish the safe profile of LENART01 regarding sedation and locomotor activity following s.c. administration in mice.
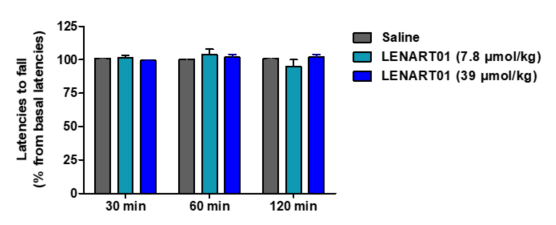
Figure 5. Effect of LENART01 in the rotarod test after s.c. administration in mice. Groups of mice received s.c. saline (control) or different doses of LENART01 (7.8 and 39 µmol/kg), and latencies to fall from the rotarod were measured before and after drug administration at different times. Latencies to fall are expressed as percentage (%) from baseline. Values represent means ± SEM (n = 5 mice per group).
Physical Dependence
The long-term use of traditional opioids is associated with physical dependence [5]. Such dependence can be visualized in opioid-dependent mice by injecting naloxone and the occurrence of withdrawal symptoms, i.e., jumps, paw tremors, head shakes and diarrhea [73][74]. To evaluate the behavioral effects of LENAR01 in mice after chronic s.c. treatment, the potential for physical dependence was determined using naloxone-precipitated withdrawal syndrome [60]. Mice were treated twice daily over a 5-day period with LENAR01 or saline (control). Administration of naloxone, two hours after the last s.c. injection of LENART01 did not induce withdrawal signs when compared to the saline-treated mice (Figure 6). Chronic administration of LENART01 did not have any significant effect on the body weights of mice compared to the saline-treated animals (Figure 7). These behavioral data indicate that the lack of LENART01 to induce withdrawal syndrome may be associated with its ability to affect D2R gene expression and due to its D2R agonist activity.
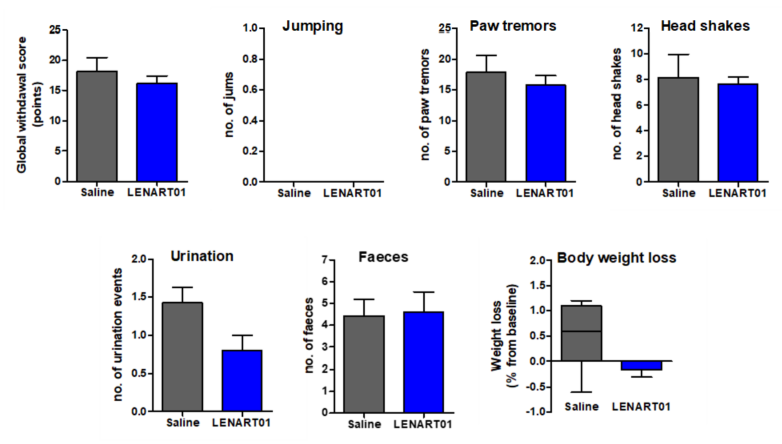
Figure 6. Withdrawal syndrome precipitated by naloxone after repeated s.c. treatment in mice with LENART01. Naloxone-induced withdrawal signs were assessed in mice that were treated twice daily for 5 days with saline (control) or LENART01 (7.8 µmol/kg). Withdrawal was precipitated two hours after last drug administration using naloxone (1 mg/kg, s.c.), and signs of withdrawal were counted over 15 min immediately after naloxone, and a global withdrawal score was calculated. Values represent means ± SEM (n = 5–7 mice per group).
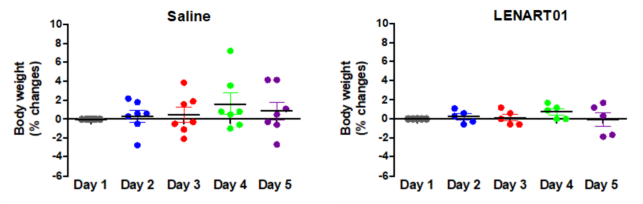
Figure 7. Weight changes in mice after chronic treatment with LENART01. Groups of mice received s.c. saline (control) or 7.8 mg/kg of LENART01 twice daily for 5 days, and changes in body weight were measured daily and are expressed as percentage (%) change from day 1. Values represent means ± SEM (n = 5–7 mice per group).
4. Conclusions
Bifunctional ligands that simultaneously target opioid receptors along with other neurotransmitter systems involved in pain modulation and opioid-induced side effects currently hold substantial scientific and clinical interest. The in vitro and in vivo pharmacology of the first dual MOR–dopamine D2R hybrid peptide, LENART01, which was designed by combining dermorphin and ranatensin pharmacophores is established. The in vitro binding studies showed LENART01 to display selectivity and full agonist activity to the human MOR. In the in vivo study, LENART01 produced potent antinociceptive effects in a model of inflammatory pain after s.c. administration in mice. Receptor antagonist in vivo studies established LENART01-induced antinociception to be mediated via MOR activation. Furthermore, this hybrid peptide exhibited higher antinociception potency compared to morphine and reduced MOR-mediated liabilities of physical dependence and sedation/motor dysfunction, thus demonstrating a better tolerability profile. Notably, LENART01 is the first dual MOR–dopamine D2R ligand investigated in vivo, and it demonstrated antinociceptive efficacy in mice after s.c. administration with reduced adverse effects of conventional opioids (Figure 8). Furthermore, LENART01 is the first peptide-based MOR-D2R ligand known to date.
The in vivo data indicate that LENART01’s activity as an MOR agonist may contribute to the beneficial effect of antinociception, while the dopamine D2R agonism may be favorable for the improved side effect profile as regards physical dependence. The current results, together with the other report [46], reveal LENART01 as a ligand with MOR and D2R agonist activity. The LENART01’s pharmacology might be supported by the molecular model findings [46], with its binding mode and interaction mechanisms with the two targets, the MOR and D2R. A bivalent peptide-based drug design to engage both the MOR and dopamine D2R may represent a promising strategy in the pursuit of a novel class of opioid analgesics devoid of opioid liabilities.
Figure 8. LENART01, a dual MOR–dopamine D2R ligand, shows antinociceptive efficacy and reduced side effects.
References
- 1. Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of chronic pain in Europe: Prevalence, impact on dai-ly life, and treatment. Pain 2006, 10, 287–333.
- Kapur, B.M.; Lala, P.K.; Shaw, J.L. Pharmacogenetics of chronic pain management. Clin. Biochem. 2014, 47, 1169–1187.
- Volkow, N.; Benveniste, H.; McLellan, T.A. Use and misuse of opioids in chronic pain. Annu. Rev. Med. 2018, 69, 451–465.
- Sobczak, Ł.; Goryński, K. Pharmacological aspects of over-the-counter opioid drugs misuse. Molecules 2020, 25, 3905.
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid analge-sia and opioid-induced adverse effects: A review. Pharmaceuticals 2021, 14, 1091.
- National Institute on Drug Abuse (NIDA). Available online: https://nida.nih.gov/research-topics/trends-statistics/overdose-death-rates (accessed on 28 February 2024).
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Annu. Rev. Neurosci. 2018, 41, 453–473.
- Pasternak, G.W. Mu opioid pharmacology: 40 years to the promised land. Adv. Pharmacol. 2018, 82, 261–291.
- Albert-Vartanian, A.; Boyd, M.; Hall, A.; Morgado, S.; Nguyen, E.; Nguyen, V.; Patel, S.; Russo, L.; Shao, A.; Raffa, R. Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse po-tential? J. Clin. Pharm. Ther. 2016, 41, 371–382.
- Busserolles, J.; Lolignier, S.; Kerckhove, N.; Bertin, C.; Authier, N.; Eschalier, A. Replacement of current opioid drugs focusing on MOR-related strategies. Pharmacol. Ther. 2020, 210, 107519.
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257.
- Che, T.; Roth, B.L. Structural insights accelerate the discovery of opioid alternatives. Annu. Rev. Biochem. 2021, 90, 739–761.
- Obeng, S.; Hiranita, T.; León, F.; McMahon, L.R.; McCurdy, C.R. Novel approaches, drug candidates, and targets in pain drug discovery. J. Med. Chem. 2021, 64, 6523–6548.
- French, A.R.; van Rijn, R.M. An updated assessment of the translational promise of G-protein-biased kappa opioid receptor agonists to treat pain and other indications without debilitating adverse effects. Pharmacol. Res. 2022, 177, 106091.
- He, Y.; Su, Q.; Zhao, L.; Zhang, L.; Yu, L.; Shi, J. Historical perspectives and recent advances in small molecule ligands of se-lective/biased/multi-targeted μ/δ/κ opioid receptor (2019–2022). Bioorg Chem. 2023, 141, 106869.
- Santino, F.; Gentilucci, L. Design of κ-opioid receptor agonists for the development of potential treatments of pain with reduced side effects. Molecules 2023, 28, 346.
- Schmidhammer, H.; Al-Khrasani, M.; Fürst, S.; Spetea, M. Peripheralization strategies applied to morphinans and implications for improved treatment of pain. Molecules 2023, 28, 4761.
- Turnaturi, R.; Piana, S.; Spoto, S.; Costanzo, G.; Reina, L.; Pasquinucci, L.; Parenti, C. From plant to chemistry: Sources of active opioid antinociceptive principles for medicinal chemistry and drug design. Molecules 2023, 28, 7089.
- Varga, B.R.; Streicher, J.M.; Majumdar, S. Strategies towards safer opioid analgesics—A review of old and upcoming targets. Br. J. Pharmacol. 2023, 180, 975–993.
- Kleczkowska, P.; Lipkowski, A.W.; Tourwé, D.; Ballet, S. Hybrid opioid/non-opioid ligands in pain research. Curr. Pharm. Des. 2013, 19, 7435–7450.
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228.
- Kleczkowska P, Nowicka K, Bujalska-Zadrozny, M., Hermans, E. Neurokinin-1 receptor-based bivalent drugs in pain man-agement: The journey to nowhere? Pharmacol. Ther. 2019, 196, 44–58.
- Starnowska-Sokół, J.; Przewłocka, B. Multifunctional opioid-derived hybrids in neuropathic pain: Preclinical evidence, ideas and challenges. Molecules 2020, 25, 5520.
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331.
- Zhuang, T.; Xiong, J.; Hao, S.; Du, W.; Liu, Z.; Liu, B.; Zhang, G.; Chen, Y. Bifunctional μ opioid and σ1 receptor ligands as novel analgesics with reduced side effects. Eur J. Med. Chem. 2021, 223, 113658.
- Spetea, M.; Schmidhammer, H. Recent chemical and pharmacological developments on 14-oxygenated-N-methylmorphinan-6- ones. Molecules 2021, 26, 5677.
- Smith, M.T.; Kong, D.; Kuo, A.; Imam, M.Z.; Williams, C.M. Multitargeted opioid ligand discovery as a strategy to retain an-algesia and reduce opioid-related adverse effects. J. Med. Chem. 2023, 66, 3746–3784.
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514.
- Listos, J.; Łupina, M.; Talarek, S.; Mazur, A.; Orzelska-Górka, J.; Kotlińska, J. The mechanisms involved in morphine addiction: An overview. Int. J. Mol. Sci. 2019, 20, 4302.
- Wise, R.A.; Robble, M.A. Dopamine and addiction. Annu. Rev. Psychol. 2020, 71, 79–106.
- Shippenberg, T.S.; Bals-Kubik, R.; Herz, A. Examination of the neurochemical substrates mediating the motivational effects of opioids: Role of the mesolimbic dopamine system and D-1 vs. D-2 dopamine receptors. J. Pharmacol. Exp. Ther. 1993, 265, 53–59.
- Gerrits, M.A.; Ramsey, N.F.; Wolterink, G.; van Ree, J.M. Lack of evidence for an involvement of nucleus accumbens dopamine D1 receptors in the initiation of heroin self-administration in the rat. Psychopharmacology 1994, 114, 486–494.
- Maldonado, R.; Saiardi, A.; Valverde, O.; Samad, T.A. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature 1997, 388, 586–589.
- Dockstader, C.; Rubinstein, M.; Grandy, D.K.; Low, M.J.; van der Kooy, D. The D2 receptor is critical in mediating opiate motivation only in opiate‐dependent and withdrawn mice. Eur. J. Neurosci. 2001, 13, 995–1001.
- Rowlett, J.K.; Platt, D.M.; Yao, W.D.; Spealman, R.D. Modulation of heroin and cocaine self-administration by dopamine D1- and D2-like receptor agonists in rhesus monkeys. J. Pharmacol. Exp. Ther. 2007, 321, 1135–1143.
- Jordan, C.J.; Humburg, B.; Rice, M.; Bi, G.-H.; You, Z.-B.; Shaik, A.B.; Cao, J.; Bonifazi, A.; Gadiano, A.; Rais, R.; et al. Newman, A.H.; Xi, Z.-X. The highly selective dopamine D3R antagonist, R-VK4-40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 2019, 158, 107597.
- You, Z.-B.; Bi, G.-H.; Galaj, E.; Kumar, V.; Cao, J.; Gadiano, A.; Rais, R.; Slusher, B.S.; Gardner, E.L.; Xi, Z.-X.; et al. Dopamine D3R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424.
- Rodgers, H.M.; Lim, S.A.; Yow, J.; Dinkins, M.L.; Patton, R.; Clemens, S.; Brewer, K.L. Dopamine D1 or D3 receptor modulators prevent morphine tolerance and reduce opioid withdrawal symptoms. Pharmacol. Biochem. Behav. 2020, 194, 172935.
- Bonifazi, A.; Battiti, F.O.; Sanchez, J.; Zaidi, S.A.; Bow, E.; Makarova, M.; Cao, J.; Shaik, A.B.; Sulima, A.; Rice, K.C.; et al. Novel dual-target μ-opioid receptor and dopamine D3 receptor ligands as potential nonaddictive pharmacotherapeutics for pain management. J. Med. Chem. 2021, 64, 7778–7808.
- Bonifazi, A.; Saab, E.; Sanchez, J.; Nazarova, A.L.; Zaidi, S.A.; Jahan, K.; Katritch, V.; Canals, M.; Lane, J.R.; Newman, A.H. Pharmacological and physicochemical properties optimization for dual-target dopamine D3 (D3R) and μ-opioid (MOR) receptor ligands as potentially safer analgesics. J. Med. Chem. 2023, 66, 10304–10341.
- Aldrich, J.V.; McLaughlin, J.P. Opioid peptides: Potential for drug development. Drug Discov. Today Technol. 2012, 9, e23–e31.
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug. Des. 2013, 81, 136–147.
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive peptides: Synthesis, sources, applications, and proposed mechanisms of action. Int. J. Mol. Sci. 2022, 23, 1445.
- Lee, Y.S. Peptidomimetics and their applications for opioid peptide drug discovery. Biomolecules 2022, 12, 1241.
- Karami Fath, M.; Babakhaniyan, K.; Zokaei, M.; Yaghoubian, A.; Akbari, S.; Khorsandi, M.; Soofi, A.; Nabi-Afjadi, M.; Zalpoor, H.; Jalalifar, F.; et al. Anti-cancer peptide-based therapeutic strategies in solid tumors. Cell Mol. Biol. Lett. 2022, 27, 33.
- Serafin, P.; Szeleszczuk, Ł.; Zhukov, I.; Szűcs, E.; Gombos, D.; Stefanucci, A.; Mollica, A.; Pisklak, D.M.; Kleczkowska, P. Opioid/dopamine receptor binding studies, NMR and molecular dynamics simulation of LENART01 chimera, an opi-oid-bombesin-like peptide. Molecules 2024, 29, 272.
- Montecucchi, P.C.; Castiglione, R. de.; Piani, S.; Gozzini, L.; Erspamer, V. Amino acid composition and sequence of dermorphin, a novel opiate-like peptide from the skin of Phyllomedusa sauvagei. Int. J. Pept. Protein Res. 1981, 17, 275–283.
- Negri, L.; Melchiorri, P.; Lattanzi, R. Pharmacology of amphibian opiate peptides. Peptides 2000, 21, 1639–1647.
- Schiller, P.W. Opioid peptide-derived analgesics. AAPS J. 2005, 7, E560–E565.
- Mizoguchi, H.; Bagetta, G.; Sakurada, T.; Sakurada, S. Dermorphin tetrapeptide analogs as potent and long-lasting analgesics with pharmacological profiles distinct from morphine. Peptides 2011, 32, 421–427.
- Hesselink, J.M.K.; Schatman, M.E. Rediscovery of old drugs: The forgotten case of dermorphin for postoperative pain and palliation. J. Pain Res. 2018, 11, 2991–2995.
- Basso N.; Marcelli M.; Ginaldi A; de Marco, M. Intrathecal dermorphine in postoperative analgesia. Peptides 1985, 6, 177–179.
- Nakajima, T.; Tanimura, T.; Pisano, J.J. Isolation and structure of a new vasoactive polypeptide. Fed. Proced. 1970, 29, 282.
- Geller, R.G.; Govier, W.C.; Pisano, J.J.; Tanimura, T.; van Clineschmidt, B. The action of ranatensin, a new polypeptide from amphibian skin, on the blood pressure of experimental animals. Br. J. Pharmacol. 1970, 40, 605-616.
- Clineschmidt, B.V.; Geller, R.G.; Govier, W.C.; Pisano, J.J.; Tanimura, T. Effects of ranatensin, a polypeptide from frog skin on isolated smooth muscle. Br. J. Pharmacol. 1971, 41, 622–628.
- Serafin, P.; Kleczkowska, P. Bombesins: A new frontier in hybrid compound development. Pharmaceutics 2023, 15, 2597.
- Zhu, X.Z.; Ji, X.Q.; Wu, S.X.; Zou, G. Sulpiride attenuates ranatensin-M-induced antinociception. Acta Pharmacol. Sinica 1991, 12, 291–293.
- Laskowska, A.K.; Szudzik, M.; Ścieżyńska, A.; Komorowski, M.; Szűcs, E.; Gombos, D.; Bączek, B.; Lipka-Miciuk, J.; Benyhe, S.; Kleczkowska, P. The role of a natural amphibian skin-based peptide, ranatensin, in pancreatic cancer expressing dopamine D2 receptors. Cancers 2022, 14, 5535.
- Serafin, P.; Kowalczyk, P.; Mollica, A.; Stefanucci, A.; Laskowska, A.K.; Zawadzka, M.; Kramkowski, K.; Kleczkowska, P. Evaluation of antimicrobial activities against various E. coli Strains of a novel hybrid peptide—LENART01. Molecules 2023, 28, 4955.
- Dumitrascuta, M.; Bermudez, M.; Trovato, O.; De Neve, J.; Ballet, S.; Wolber, G.; Spetea, M. Antinociceptive efficacy of the µ-opioid/nociceptin peptide-based hybrid KGNOP1 in inflammatory pain without rewarding effects in mice: An experimental assessment and molecular docking. Molecules 2021, 26, 3267.
- Amiche, M.; Sagan, S.; Mor, A.; Pelaprat, D.; Rostene, W.; Delfour, A.; Nicolas, P. Characterisation and visualisation of 3Hdermorphin binding to mu opioid receptors in the rat brain. Combined high selectivity and affinity in a natural peptide agonist for the morphine (mu) receptor. Eur. J. Biochem. 1990, 189, 625–635.
- Negri, L.; Erspamer, G.F.; Severini, C.; Potenza, R.L.; Melchiorri, P.; Erspamer, V. Dermorphin-related peptides from the skin of Phyllomedusa bicolor and their amidated analogs activate two mu opioid receptor subtypes that modulate antinociception and catalepsy in the rat. Proc. Natl. Acad. Sci. USA 1992, 89, 7203–7207.
- Bird, M.F.; Cerlesi, M.C.; Brown, M.; Malfacini, D.; Vezzi, V.; Molinari, P.; Micheli, L.; Di Cesare Mannelli, L.; Ghelardini, C.; Guerrini, R.; et al Characterisation of the novel mixed Mu-NOP peptide ligand dermorphin-N/OFQ (DeNo). PLoS ONE 2016, 11, e0156897.
- Giakomidi, D.; Bird, M.F.; McDonald, J.; Marzola, E.; Guerrini, R.; Chanoch, S.; Sabu, N.; Horley, B.; Calo, G.; Lambert, D.G. Evaluation of Cys(ATTO 488)8Dermorphin-NH2 as a novel tool for the study of μ-opioid peptide receptors. PLoS ONE 2021, 16, e0250011.
- D’Amour, F.E.; Smith, D.L. A method for determining loss of pain sensation, J. Pharmacol. Exp. Ther. 1941, 72, 74–79.
- Dumitrascuta, M.; Bermudez, M.; Ben Haddou, T.; Guerrieri, E.; Schläfer, L.; Ritsch, A.; Hosztafi, S.; Lantero, A.; Kreutz, C.; Massotte, D.; et al. N-Phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones turns selective µ opioid receptor lig-ands into dual µ/δ opioid receptor agonists. Sci. Rep. 2020, 10, 5653.
- Meert, T.F.; Vermeirsch, H.A. A preclinical comparison between different opioids: Antinociceptive versus adverse effects. Pharmacol. Biochem. Behav. 2005, 80, 309–326.
- Spetea, M.; Bohotin, C.R.; Asim, M.F.; Stübegger, K.; Schmidhammer, H. In vitro and in vivo pharmacological profile of the 5-benzyl analogue of 14-methoxymetopon, a novel mu opioid analgesic with reduced propensity to alter motor function. Eur. J. Pharm. Sci. 2010, 41, 125–135.
- Ben Haddou, T.; Béni, S.; Hosztafi, S.; Malfacini, D.; Calo, G.; Schmidhammer, H.; Spetea, M. Pharmacological investigations of N-substituent variation in morphine and oxymorphone: Opioid receptor binding, signaling and antinociceptive activity. PLoS ONE 2014, 9, e99231.
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652.
- Dubuisson, D.; Dennis, S.G. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174.
- Jones, B.J.; Roberts, D.J. The quantitative measurement of motor inco-ordination in naive mice using an accelerating rotarod. J. Pharm. Pharmacol. 1968, 20, 302–304.
- Buckett, W.R. A new test for morphine-like physical dependence (addiction liability) in rats. Psychopharmacologia 1964, 6, 410–416.
- Matthes, H.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the µ-opioid-receptor gene. Nature 1996, 383, 819–823.

