 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sona Ciernikova | -- | 3316 | 2024-04-09 18:43:42 |
Video Upload Options
DNA methylation represents a crucial mechanism of epigenetic regulation in hematologic malignancies. The methylation process is controlled by specific DNA methyl transferases and other regulators, which are often affected by genetic alterations. Global hypomethylation and hypermethylation of tumor suppressor genes are associated with hematologic cancer development and progression. Several epi-drugs have been successfully implicated in the treatment of hematologic malignancies, including the hypomethylating agents (HMAs) decitabine and azacytidine. However, combinations with other treatment modalities and the discovery of new molecules are still the subject of research to increase sensitivity to anti-cancer therapies and improve patient outcomes.
1. Introduction
2. DNA Methylation of Target Genes in Leukemias, Myelodysplastic Syndromes, and Lymphomas
2.1. Aberrant Methylation in MDS and AML
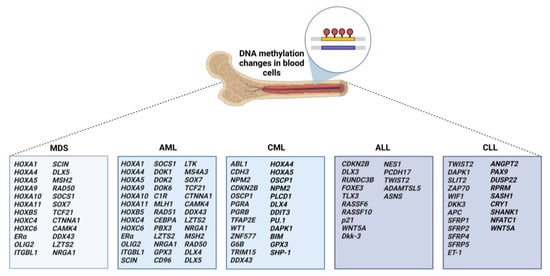
2.2. Aberrant Methylation in CML
2.3. Aberrant Methylation in ALL
2.4. Aberrant Methylation in CLL
2.5. Aberrant Methylation in Malignant Lymphomas
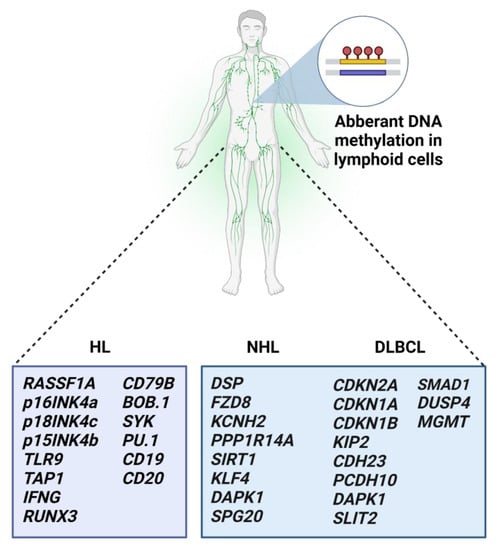
In conclusion, DNA methylation is one of the main epigenetic mechanisms, besides known genetic alterations, that play a role in cancer initiation and progression. In the future, DNA methylation-based stratification of hematologic patients might lead to more personalized treatment with better outcomes. The reversibility of changes in DNA methylation landscapes enables broad clinical implications. However, adverse events associated with indiscriminate global hypomethylation with DNA methylation inhibitors are a matter of concern, and further investigations are highly warranted. According to the findings, DNA methylation processes in hematologic malignancies are usually associated with other mechanisms of epigenetic regulations, including histone modifications and miRNA regulation. Thus, evaluating the role of epigenetic modifications in a more complex matter would be beneficial for blood cancer patients.
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638.
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Weisenberger, D.J.; Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation and DNA Methyltransferases in Cancer. Adv. Exp. Med. Biol. 2022, 1389, 317–348.
- Dan, J.; Chen, T. Genetic Studies on Mammalian DNA Methyltransferases. Adv. Exp. Med. Biol. 2022, 1389, 111–136.
- Cypris, O.; Frobel, J.; Rai, S.; Franzen, J.; Sontag, S.; Goetzke, R.; de Toledo, M.A.S.; Zenke, M.; Wagner, W. Tracking of epigenetic changes during hematopoietic differentiation of induced pluripotent stem cells. Clin. Epigenet. 2019, 11, 19.
- Farlik, M.; Halbritter, F.; Muller, F.; Choudry, F.A.; Ebert, P.; Klughammer, J.; Farrow, S.; Santoro, A.; Ciaurro, V.; Mathur, A.; et al. DNA Methylation Dynamics of Human Hematopoietic Stem Cell Differentiation. Cell Stem Cell 2016, 19, 808–822.
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83.
- Gao, H.; He, X.; Li, Q.; Wang, Y.; Tian, Y.; Chen, X.; Wang, J.; Guo, Y.; Wang, W.; Li, X. Genome-wide DNA methylome analysis reveals methylation subtypes with different clinical outcomes for acute myeloid leukemia patients. Cancer Med. 2020, 9, 6296–6305.
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C.; et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861–1868.
- Pei, L.; Choi, J.H.; Liu, J.; Lee, E.J.; McCarthy, B.; Wilson, J.M.; Speir, E.; Awan, F.; Tae, H.; Arthur, G.; et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics 2012, 7, 567–578.
- Hetzel, S.; Mattei, A.L.; Kretzmer, H.; Qu, C.; Chen, X.; Fan, Y.; Wu, G.; Roberts, K.G.; Luger, S.; Litzow, M.; et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 2022, 3, 768–782.
- Haider, Z.; Landfors, M.; Golovleva, I.; Erlanson, M.; Schmiegelow, K.; Flaegstad, T.; Kanerva, J.; Noren-Nystrom, U.; Hultdin, M.; Degerman, S. DNA methylation and copy number variation profiling of T-cell lymphoblastic leukemia and lymphoma. Blood Cancer J. 2020, 10, 45.
- Liu, P.; Jiang, W.; Zhao, J.; Zhang, H. Integrated analysis of genome-wide gene expression and DNA methylation microarray of diffuse large B-cell lymphoma with TET mutations. Mol. Med. Rep. 2017, 16, 3777–3782.
- Roux, B.; Picou, F.; Debeissat, C.; Koubi, M.; Gallay, N.; Hirsch, P.; Ravalet, N.; Bene, M.C.; Maigre, M.; Hunault, M.; et al. Aberrant DNA methylation impacts HOX genes expression in bone marrow mesenchymal stromal cells of myelodysplastic syndromes and de novo acute myeloid leukemia. Cancer Gene Ther. 2022, 29, 1263–1275.
- Sampath, S.; Misra, P.; Yadav, S.K.; Sharma, S.; Somasundaram, V. A study on DNA methylation status in promoter region of p15 gene in patients of acute myeloid leukemia and myelodysplastic syndrome. Med. J. Armed Forces India 2021, 77, 337–342.
- Fu, H.Y.; Wu, D.S.; Zhou, H.R.; Shen, J.Z. CpG island methylator phenotype and its relationship with prognosis in adult acute leukemia patients. Hematology 2014, 19, 329–337.
- Kurtovic, N.K.; Krajnovic, M.; Bogdanovic, A.; Suvajdzic, N.; Jovanovic, J.; Dimitrijevic, B.; Colovic, M.; Krtolica, K. Concomitant aberrant methylation of p15 and MGMT genes in acute myeloid leukemia: Association with a particular immunophenotype of blast cells. Med. Oncol. 2012, 29, 3547–3556.
- Hiller, J.K.; Schmoor, C.; Gaidzik, V.I.; Schmidt-Salzmann, C.; Yalcin, A.; Abdelkarim, M.; Blagitko-Dorfs, N.; Dohner, K.; Bullinger, L.; Duyster, J.; et al. Evaluating the impact of genetic and epigenetic aberrations on survival and response in acute myeloid leukemia patients receiving epigenetic therapy. Ann. Hematol. 2017, 96, 559–565.
- Lian, X.Y.; Ma, J.C.; Zhou, J.D.; Zhang, T.J.; Wu, D.H.; Deng, Z.Q.; Zhang, Z.H.; Li, X.X.; He, P.F.; Yan, Y.; et al. Hypermethylation of ITGBL1 is associated with poor prognosis in acute myeloid leukemia. J. Cell. Physiol. 2019, 234, 9438–9446.
- Zhang, Z.H.; Zhang, W.; Zhou, J.D.; Zhang, T.J.; Ma, J.C.; Xu, Z.J.; Lian, X.Y.; Wu, D.H.; Wen, X.M.; Deng, Z.Q.; et al. Decreased SCIN expression, associated with promoter methylation, is a valuable predictor for prognosis in acute myeloid leukemia. Mol. Carcinog. 2018, 57, 735–744.
- Zhang, T.J.; Xu, Z.J.; Gu, Y.; Wen, X.M.; Ma, J.C.; Zhang, W.; Deng, Z.Q.; Leng, J.Y.; Qian, J.; Lin, J.; et al. Identification and validation of prognosis-related DLX5 methylation as an epigenetic driver in myeloid neoplasms. Clin. Transl. Med. 2020, 10, e29.
- Park, S.; So, M.K.; Huh, J. Methylation of DNA Repair Genes as a Prognostic Biomarker in AML of a TCGA-LAML Cohort. Clin. Lab. 2022, 68, 7.
- Chaubey, R.; Sazawal, S.; Mahapatra, M.; Chhikara, S.; Saxena, R. Prognostic relevance of aberrant SOCS-1 gene promoter methylation in myelodysplastic syndromes patients. Int. J. Lab. Hematol. 2015, 37, 265–271.
- He, P.F.; Xu, Z.J.; Zhou, J.D.; Li, X.X.; Zhang, W.; Wu, D.H.; Zhang, Z.H.; Lian, X.Y.; Yao, X.Y.; Deng, Z.Q.; et al. Methylation-associated DOK1 and DOK2 down-regulation: Potential biomarkers for predicting adverse prognosis in acute myeloid leukemia. J. Cell. Physiol. 2018, 233, 6604–6614.
- Sun, G.K.; Tang, L.J.; Zhou, J.D.; Xu, Z.J.; Yang, L.; Yuan, Q.; Ma, J.C.; Liu, X.H.; Lin, J.; Qian, J.; et al. DOK6 promoter methylation serves as a potential biomarker affecting prognosis in de novo acute myeloid leukemia. Cancer Med. 2019, 8, 6393–6402.
- Bozic, T.; Lin, Q.; Frobel, J.; Wilop, S.; Hoffmann, M.; Muller-Tidow, C.; Brummendorf, T.H.; Jost, E.; Wagner, W. DNA-methylation in C1R is a prognostic biomarker for acute myeloid leukemia. Clin. Epigenet. 2015, 7, 116.
- Sestakova, S.; Cerovska, E.; Salek, C.; Kundrat, D.; Jeziskova, I.; Folta, A.; Mayer, J.; Racil, Z.; Cetkovsky, P.; Remesova, H. A validation study of potential prognostic DNA methylation biomarkers in patients with acute myeloid leukemia using a custom DNA methylation sequencing panel. Clin. Epigenet. 2022, 14, 22.
- Zhou, J.D.; Yao, D.M.; Zhang, Y.Y.; Ma, J.C.; Wen, X.M.; Yang, J.; Guo, H.; Chen, Q.; Lin, J.; Qian, J. GPX3 hypermethylation serves as an independent prognostic biomarker in non-M3 acute myeloid leukemia. Am. J. Cancer Res. 2015, 5, 1786–1794.
- Zhou, J.D.; Zhang, T.J.; Wang, Y.X.; Yang, D.Q.; Yang, L.; Ma, J.C.; Wen, X.M.; Yang, J.; Lin, J.; Qian, J. DLX4 hypermethylation is a prognostically adverse indicator in de novo acute myeloid leukemia. Tumour Biol. 2016, 37, 8951–8960.
- Li, J.; Jin, W.; Tan, Y.; Wang, B.; Wang, X.; Zhao, M.; Wang, K. Distinct gene expression pattern of RUNX1 mutations coordinated by target repression and promoter hypermethylation in acute myeloid leukemia. Front. Med. 2022, 16, 627–636.
- Man, C.H.; Fung, T.K.; Wan, H.; Cher, C.Y.; Fan, A.; Ng, N.; Ho, C.; Wan, T.S.; Tanaka, T.; So, C.W.; et al. Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia. Blood 2015, 125, 3928–3936.
- Gu, Y.; Zhou, J.D.; Xu, Z.J.; Zhang, T.J.; Wen, X.M.; Ma, J.C.; Ji, R.B.; Yuan, Q.; Zhang, W.; Chen, Q.; et al. Promoter methylation of the candidate tumor suppressor gene TCF21 in myelodysplastic syndrome and acute myeloid leukemia. Am. J. Transl. Res. 2019, 11, 3450–3460.
- Chen, X.X.; Lin, J.; Qian, J.; Qian, W.; Yang, J.; Ma, J.C.; Deng, Z.Q.; An, C.; Tang, C.Y.; Qian, Z.; et al. Methylation of CTNNA1 promoter: Frequent but not an adverse prognostic factor in acute myeloid leukemia. Leuk. Res. 2014, 38, 613–618.
- Chen, W.W.; Liu, D.B.; Xiao, H.X.; Zhou, L.J.; Qu, J. Identification of differentially expressed genes induced by aberrant methylation in acute myeloid leukemia using integrated bioinformatics analyses. Oncol. Lett. 2022, 24, 383.
- Lin, J.; Chen, Q.; Yang, J.; Qian, J.; Deng, Z.Q.; Qian, W.; Chen, X.X.; Ma, J.C.; Xiong, D.S.; Ma, Y.J.; et al. DDX43 promoter is frequently hypomethylated and may predict a favorable outcome in acute myeloid leukemia. Leuk. Res. 2014, 38, 601–607.
- Qu, X.; Othus, M.; Davison, J.; Wu, Y.; Yan, L.; Meshinchi, S.; Ostronoff, F.; Estey, E.H.; Radich, J.P.; Erba, H.P.; et al. Prognostic methylation markers for overall survival in cytogenetically normal patients with acute myeloid leukemia treated on SWOG trials. Cancer 2017, 123, 2472–2481.
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117.
- Jelinek, J.; Gharibyan, V.; Estecio, M.R.; Kondo, K.; He, R.; Chung, W.; Lu, Y.; Zhang, N.; Liang, S.; Kantarjian, H.M.; et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS ONE 2011, 6, e22110.
- Maupetit-Mehouas, S.; Court, F.; Bourgne, C.; Guerci-Bresler, A.; Cony-Makhoul, P.; Johnson, H.; Etienne, G.; Rousselot, P.; Guyotat, D.; Janel, A.; et al. DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34+ CD15− cells in early chronic-phase chronic myeloid leukemia. Mol. Oncol. 2018, 12, 814–829.
- Chen, Q.; Lin, J.; Qian, J.; Deng, Z.Q.; Qian, W.; Yang, J.; Li, Y.; Chen, X.X.; Ma, Y.J.; Ma, J.C.; et al. The methylation status of the DDX43 promoter in Chinese patients with chronic myeloid leukemia. Genet. Test. Mol. Biomarkers 2013, 17, 508–511.
- Elias, M.H.; Azlan, H.; Sulong, S.; Baba, A.A.; Ankathil, R. Aberrant DNA methylation at HOXA4 and HOXA5 genes are associated with resistance to imatinib mesylate among chronic myeloid leukemia patients. Cancer Rep. 2018, 1, e1111.
- Song, J.J.; Liu, Q.; Li, Y.; Yang, Z.S.; Yang, L.; Xiang, T.X.; Ren, G.S.; Chen, J.B. Epigenetic inactivation of PLCD1 in chronic myeloid leukemia. Int. J. Mol. Med. 2012, 30, 179–184.
- Zhou, J.D.; Wang, Y.X.; Zhang, T.J.; Yang, D.Q.; Yao, D.M.; Guo, H.; Yang, L.; Ma, J.C.; Wen, X.M.; Yang, J.; et al. Epigenetic inactivation of DLX4 is associated with disease progression in chronic myeloid leukemia. Biochem. Biophys. Res. Commun. 2015, 463, 1250–1256.
- Wang, Y.L.; Qian, J.; Lin, J.; Yao, D.M.; Qian, Z.; Zhu, Z.H.; Li, J.Y. Methylation status of DDIT3 gene in chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2010, 29, 54.
- Yang, H.; Liang, H.; Yan, J.S.; Tao, R.; Hao, S.G.; Ma, L.Y. Down-regulation of hematopoiesis master regulator PU.1 via aberrant methylation in chronic myeloid leukemia. Int. J. Hematol. 2012, 96, 65–73.
- Qian, J.; Wang, Y.L.; Lin, J.; Yao, D.M.; Xu, W.R.; Wu, C.Y. Aberrant methylation of the death-associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur. J. Haematol. 2009, 82, 119–123.
- San Jose-Eneriz, E.; Agirre, X.; Jimenez-Velasco, A.; Cordeu, L.; Martin, V.; Arqueros, V.; Garate, L.; Fresquet, V.; Cervantes, F.; Martinez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 2009, 45, 1877–1889.
- Yao, D.M.; Zhou, J.D.; Zhang, Y.Y.; Yang, L.; Wen, X.M.; Yang, J.; Guo, H.; Chen, Q.; Lin, J.; Qian, J. GPX3 promoter is methylated in chronic myeloid leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 6450–6457.
- Li, Y.; Yang, L.; Pan, Y.; Yang, J.; Shang, Y.; Luo, J. Methylation and decreased expression of SHP-1 are related to disease progression in chronic myelogenous leukemia. Oncol. Rep. 2014, 31, 2438–2446.
- Wenzinger, C.; Williams, E.; Gru, A.A. Updates in the Pathology of Precursor Lymphoid Neoplasms in the Revised Fourth Edition of the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Curr. Hematol. Malig. Rep. 2018, 13, 275–288.
- Touzart, A.; Boissel, N.; Belhocine, M.; Smith, C.; Graux, C.; Latiri, M.; Lhermitte, L.; Mathieu, E.L.; Huguet, F.; Lamant, L.; et al. Low level CpG island promoter methylation predicts a poor outcome in adult T-cell acute lymphoblastic leukemia. Haematologica 2020, 105, 1575–1581.
- Jang, W.; Park, J.; Kwon, A.; Choi, H.; Kim, J.; Lee, G.D.; Han, E.; Jekarl, D.W.; Chae, H.; Han, K.; et al. CDKN2B downregulation and other genetic characteristics in T-acute lymphoblastic leukemia. Exp. Mol. Med. 2019, 51, 1–15.
- Dell’Orto, M.C.; Banelli, B.; Giarin, E.; Accordi, B.; Trentin, L.; Romani, M.; Te Kronnie, G.; Basso, G. Down-regulation of DLX3 expression in MLL-AF4 childhood lymphoblastic leukemias is mediated by promoter region hypermethylation. Oncol. Rep. 2007, 18, 417–423.
- Burmeister, D.W.; Smith, E.H.; Cristel, R.T.; McKay, S.D.; Shi, H.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. The expression of RUNDC3B is associated with promoter methylation in lymphoid malignancies. Hematol. Oncol. 2017, 35, 25–33.
- Chatterton, Z.; Burke, D.; Emslie, K.R.; Craig, J.M.; Ng, J.; Ashley, D.M.; Mechinaud, F.; Saffery, R.; Wong, N.C. Validation of DNA methylation biomarkers for diagnosis of acute lymphoblastic leukemia. Clin. Chem. 2014, 60, 995–1003.
- Younesian, S.; Shahkarami, S.; Ghaffari, P.; Alizadeh, S.; Mehrasa, R.; Ghavamzadeh, A.; Ghaffari, S.H. DNA hypermethylation of tumor suppressor genes RASSF6 and RASSF10 as independent prognostic factors in adult acute lymphoblastic leukemia. Leuk. Res. 2017, 61, 33–38.
- Younesian, S.; Shahkarami, S.; Ghaffari, P.; Alizadeh, S.; Mehrasa, R.; Ghaffari, S.H. Residual methylation of tumor suppressor gene promoters, RASSF6 and RASSF10, as novel biomarkers for minimal residual disease detection in adult acute lymphoblastic leukemia. Ann. Hematol. 2019, 98, 2719–2727.
- Roman-Gomez, J.; Castillejo, J.A.; Jimenez, A.; Gonzalez, M.G.; Moreno, F.; Rodriguez, M.C.; Barrios, M.; Maldonado, J.; Torres, A. 5’ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 2002, 99, 2291–2296.
- Roman-Gomez, J.; Jimenez-Velasco, A.; Cordeu, L.; Vilas-Zornoza, A.; San Jose-Eneriz, E.; Garate, L.; Castillejo, J.A.; Martin, V.; Prosper, F.; Heiniger, A.; et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur. J. Cancer 2007, 43, 2736–2746.
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.A.; Navarro, G.; Barrios, M.; Andreu, E.J.; Prosper, F.; Heiniger, A.; Torres, A. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br. J. Cancer 2004, 91, 707–713.
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.A.; Barrios, M.; Andreu, E.J.; Prosper, F.; Heiniger, A.; Torres, A. The normal epithelial cell-specific 1 (NES1) gene, a candidate tumor suppressor gene on chromosome 19q13.3-4, is downregulated by hypermethylation in acute lymphoblastic leukemia. Leukemia 2004, 18, 362–365.
- Narayan, G.; Freddy, A.J.; Xie, D.; Liyanage, H.; Clark, L.; Kisselev, S.; Un Kang, J.; Nandula, S.V.; McGuinn, C.; Subramaniyam, S.; et al. Promoter methylation-mediated inactivation of PCDH10 in acute lymphoblastic leukemia contributes to chemotherapy resistance. Genes Chromosomes Cancer 2011, 50, 1043–1053.
- Uyen, T.N.; Sakashita, K.; Al-Kzayer, L.F.; Nakazawa, Y.; Kurata, T.; Koike, K. Aberrant methylation of protocadherin 17 and its prognostic value in pediatric acute lymphoblastic leukemia. Pediatr. Blood Cancer 2017, 64, e26259.
- Thathia, S.H.; Ferguson, S.; Gautrey, H.E.; van Otterdijk, S.D.; Hili, M.; Rand, V.; Moorman, A.V.; Meyer, S.; Brown, R.; Strathdee, G. Epigenetic inactivation of TWIST2 in acute lymphoblastic leukemia modulates proliferation, cell survival and chemosensitivity. Haematologica 2012, 97, 371–378.
- Abdullah, M.; Choo, C.W.; Alias, H.; Rahman, E.J.A.; Ibrahim, H.M.; Jamal, R.; Hussin, N.H. ADAMTSL5 and CDH11: Putative epigenetic markers for therapeutic resistance in acute lymphoblastic leukemia. Hematology 2017, 22, 386–391.
- Akahane, K.; Kimura, S.; Miyake, K.; Watanabe, A.; Kagami, K.; Yoshimura, K.; Shinohara, T.; Harama, D.; Kasai, S.; Goi, K.; et al. Association of allele-specific methylation of the ASNS gene with asparaginase sensitivity and prognosis in T-ALL. Blood Adv. 2022, 6, 212–224.
- Hallek, M.; Shanafelt, T.D.; Eichhorst, B. Chronic lymphocytic leukaemia. Lancet 2018, 391, 1524–1537.
- Cahill, N.; Rosenquist, R. Uncovering the DNA methylome in chronic lymphocytic leukemia. Epigenetics 2013, 8, 138–148.
- Raval, A.; Lucas, D.M.; Matkovic, J.J.; Bennett, K.L.; Liyanarachchi, S.; Young, D.C.; Rassenti, L.; Kipps, T.J.; Grever, M.R.; Byrd, J.C.; et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J. Clin. Oncol. 2005, 23, 3877–3885.
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890.
- Dunwell, T.L.; Dickinson, R.E.; Stankovic, T.; Dallol, A.; Weston, V.; Austen, B.; Catchpoole, D.; Maher, E.R.; Latif, F. Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics 2009, 4, 265–269.
- Claus, R.; Lucas, D.M.; Stilgenbauer, S.; Ruppert, A.S.; Yu, L.; Zucknick, M.; Mertens, D.; Buhler, A.; Oakes, C.C.; Larson, R.A.; et al. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J. Clin. Oncol. 2012, 30, 2483–2491.
- Chim, C.S.; Pang, R.; Liang, R. Epigenetic dysregulation of the Wnt signalling pathway in chronic lymphocytic leukaemia. J. Clin. Pathol. 2008, 61, 1214–1219.
- Bennett, L.B.; Taylor, K.H.; Arthur, G.L.; Rahmatpanah, F.B.; Hooshmand, S.I.; Caldwell, C.W. Epigenetic regulation of WNT signaling in chronic lymphocytic leukemia. Epigenomics 2010, 2, 53–70.
- Martinelli, S.; Maffei, R.; Fiorcari, S.; Quadrelli, C.; Zucchini, P.; Benatti, S.; Potenza, L.; Luppi, M.; Marasca, R. The expression of endothelin-1 in chronic lymphocytic leukemia is controlled by epigenetic mechanisms and extracellular stimuli. Leuk. Res. 2017, 54, 17–24.
- Martinelli, S.; Kanduri, M.; Maffei, R.; Fiorcari, S.; Bulgarelli, J.; Marasca, R.; Rosenquist, R. ANGPT2 promoter methylation is strongly associated with gene expression and prognosis in chronic lymphocytic leukemia. Epigenetics 2013, 8, 720–729.
- Rani, L.; Mathur, N.; Gupta, R.; Gogia, A.; Kaur, G.; Dhanjal, J.K.; Sundar, D.; Kumar, L.; Sharma, A. Genome-wide DNA methylation profiling integrated with gene expression profiling identifies PAX9 as a novel prognostic marker in chronic lymphocytic leukemia. Clin. Epigenet. 2017, 9, 57.
- Pan, H.; Renaud, L.; Chaligne, R.; Bloehdorn, J.; Tausch, E.; Mertens, D.; Fink, A.M.; Fischer, K.; Zhang, C.; Betel, D.; et al. Discovery of Candidate DNA Methylation Cancer Driver Genes. Cancer Discov. 2021, 11, 2266–2281.
- Hanoun, M.; Eisele, L.; Suzuki, M.; Greally, J.M.; Huttmann, A.; Aydin, S.; Scholtysik, R.; Klein-Hitpass, L.; Duhrsen, U.; Durig, J. Epigenetic silencing of the circadian clock gene CRY1 is associated with an indolent clinical course in chronic lymphocytic leukemia. PLoS ONE 2012, 7, e34347.
- Loi, E.; Moi, L.; Fadda, A.; Satta, G.; Zucca, M.; Sanna, S.; Amini Nia, S.; Cabras, G.; Padoan, M.; Magnani, C.; et al. Methylation alteration of SHANK1 as a predictive, diagnostic and prognostic biomarker for chronic lymphocytic leukemia. Oncotarget 2019, 10, 4987–5002.
- Wolf, C.; Garding, A.; Filarsky, K.; Bahlo, J.; Robrecht, S.; Becker, N.; Zucknick, M.; Rouhi, A.; Weigel, A.; Claus, R.; et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int. J. Cancer 2018, 142, 322–333.
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162.
- Kanduri, M.; Cahill, N.; Goransson, H.; Enstrom, C.; Ryan, F.; Isaksson, A.; Rosenquist, R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010, 115, 296–305.
- Poppova, L.; Pavlova, S.; Gonzalez, B.; Kotaskova, J.; Plevova, K.; Dumbovic, G.; Janovska, P.; Bystry, V.; Panovska, A.; Bezdekova, L.; et al. Memory B-cell like chronic lymphocytic leukaemia is associated with specific methylation profile of WNT5A promoter and undetectable expression of WNT5A gene. Epigenetics 2022, 17, 1628–1635.
- Rodriguez-Abreu, D.; Bordoni, A.; Zucca, E. Epidemiology of hematological malignancies. Ann. Oncol. 2007, 18 (Suppl. S1), i3–i8.
- Murray, P.G.; Qiu, G.H.; Fu, L.; Waites, E.R.; Srivastava, G.; Heys, D.; Agathanggelou, A.; Latif, F.; Grundy, R.G.; Mann, J.R.; et al. Frequent epigenetic inactivation of the RASSF1A tumor suppressor gene in Hodgkin’s lymphoma. Oncogene 2004, 23, 1326–1331.
- Garcia, M.J.; Martinez-Delgado, B.; Cebrian, A.; Martinez, A.; Benitez, J.; Rivas, C. Different incidence and pattern of p15INK4b and p16INK4a promoter region hypermethylation in Hodgkin’s and CD30-Positive non-Hodgkin’s lymphomas. Am. J. Pathol. 2002, 161, 1007–1013.
- Ushmorov, A.; Leithauser, F.; Sakk, O.; Weinhausel, A.; Popov, S.W.; Moller, P.; Wirth, T. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood 2006, 107, 2493–2500.
- Xia, C.; Olsen, T.K.; Zirakzadeh, A.A.; Almamoun, R.; Sjoholm, L.K.; Dahlstrom, J.; Sjoberg, J.; Claesson, H.E.; Johnsen, J.I.; Winqvist, O.; et al. Hodgkin Lymphoma Monozygotic Triplets Reveal Divergences in DNA Methylation Signatures. Front. Oncol. 2020, 10, 598872.
- Bethge, N.; Honne, H.; Hilden, V.; Troen, G.; Eknaes, M.; Liestol, K.; Holte, H.; Delabie, J.; Smeland, E.B.; Lind, G.E. Identification of highly methylated genes across various types of B-cell non-hodgkin lymphoma. PLoS ONE 2013, 8, e79602.
- Bethge, N.; Honne, H.; Andresen, K.; Hilden, V.; Troen, G.; Liestol, K.; Holte, H.; Delabie, J.; Lind, G.E.; Smeland, E.B. A gene panel, including LRP12, is frequently hypermethylated in major types of B-cell lymphoma. PLoS ONE 2014, 9, e104249.
- Frazzi, R.; Zanetti, E.; Pistoni, M.; Tamagnini, I.; Valli, R.; Braglia, L.; Merli, F. Methylation changes of SIRT1, KLF4, DAPK1 and SPG20 in B-lymphocytes derived from follicular and diffuse large B-cell lymphoma. Leuk. Res. 2017, 57, 89–96.
- Frazzi, R.; Cusenza, V.Y.; Pistoni, M.; Canovi, L.; Cascione, L.; Bertoni, F.; Merli, F. KLF4, DAPK1 and SPG20 promoter methylation is not affected by DNMT1 silencing and hypomethylating drugs in lymphoma cells. Oncol. Rep. 2022, 47, 10.
- Chambwe, N.; Kormaksson, M.; Geng, H.; De, S.; Michor, F.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Godley, L.A.; Gascoyne, R.D.; et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 2014, 123, 1699–1708.
- Zainuddin, N.; Kanduri, M.; Berglund, M.; Lindell, M.; Amini, R.M.; Roos, G.; Sundstrom, C.; Enblad, G.; Rosenquist, R. Quantitative evaluation of p16(INK4a) promoter methylation using pyrosequencing in de novo diffuse large B-cell lymphoma. Leuk. Res. 2011, 35, 438–443.
- Hagiwara, K.; Li, Y.; Kinoshita, T.; Kunishma, S.; Ohashi, H.; Hotta, T.; Nagai, H. Aberrant DNA methylation of the p57KIP2 gene is a sensitive biomarker for detecting minimal residual disease in diffuse large B cell lymphoma. Leuk. Res. 2010, 34, 50–54.
- Shawky, S.A.; El-Borai, M.H.; Khaled, H.M.; Guda, I.; Mohanad, M.; Abdellateif, M.S.; Zekri, A.N.; Bahanasy, A.A. The prognostic impact of hypermethylation for a panel of tumor suppressor genes and cell of origin subtype on diffuse large B-cell lymphoma. Mol. Biol. Rep. 2019, 46, 4063–4076.
- Cao, B.; Guo, X.; Huang, L.; Wang, B.; Wang, W.; Han, D.; Zhang, W.; Zhong, K. Methylation silencing CDH23 is a poor prognostic marker in diffuse large B-cell lymphoma. Aging 2021, 13, 17768–17788.
- Huang, W.; Xue, X.; Shan, L.; Qiu, T.; Guo, L.; Ying, J.; Lu, N. Clinical significance of PCDH10 promoter methylation in diffuse large B-cell lymphoma. BMC Cancer 2017, 17, 815.
- Kristensen, L.S.; Asmar, F.; Dimopoulos, K.; Nygaard, M.K.; Aslan, D.; Hansen, J.W.; Ralfkiaer, E.; Gronbaek, K. Hypermethylation of DAPK1 is an independent prognostic factor predicting survival in diffuse large B-cell lymphoma. Oncotarget 2014, 5, 9798–9810.
- Wang, H.; Zhou, L.Y.; Guan, Z.B.; Zeng, W.B.; Zhou, L.L.; Liu, Y.N.; Pan, X.Y. Prognostic significance of DAPK promoter methylation in lymphoma: A meta-analysis. PLoS ONE 2019, 14, e0210943.
- Kristensen, L.S.; Hansen, J.W.; Kristensen, S.S.; Tholstrup, D.; Harslof, L.B.; Pedersen, O.B.; De Nully Brown, P.; Gronbaek, K. Aberrant methylation of cell-free circulating DNA in plasma predicts poor outcome in diffuse large B cell lymphoma. Clin. Epigenet. 2016, 8, 95.
- Mohamed, G.; Talima, S.; Li, L.; Wei, W.; Rudzki, Z.; Allam, R.M.; Simmons, W.; Tao, Q.; Murray, P.G. Low Expression and Promoter Hypermethylation of the Tumour Suppressor SLIT2, are Associated with Adverse Patient Outcomes in Diffuse Large B Cell Lymphoma. Pathol. Oncol. Res. 2019, 25, 1223–1231.
- Schmid, C.A.; Robinson, M.D.; Scheifinger, N.A.; Muller, S.; Cogliatti, S.; Tzankov, A.; Muller, A. DUSP4 deficiency caused by promoter hypermethylation drives JNK signaling and tumor cell survival in diffuse large B cell lymphoma. J. Exp. Med. 2015, 212, 775–792.
- Lee, S.M.; Lee, E.J.; Ko, Y.H.; Lee, S.H.; Maeng, L.; Kim, K.M. Prognostic significance of O6-methylguanine DNA methyltransferase and p57 methylation in patients with diffuse large B-cell lymphomas. APMIS 2009, 117, 87–94.
- Clozel, T.; Yang, S.; Elstrom, R.L.; Tam, W.; Martin, P.; Kormaksson, M.; Banerjee, S.; Vasanthakumar, A.; Culjkovic, B.; Scott, D.W.; et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013, 3, 1002–1019.

