 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Heba Abdel-Mohsen | -- | 1649 | 2024-03-25 01:44:23 | | | |
| 2 | Jessie Wu | Meta information modification | 1649 | 2024-03-25 06:11:06 | | |
Video Upload Options
Protein kinases are one of the largest enzyme families. By manipulating the location, activity, and functionality of many proteins via multisite phosphorylation, they regulate a broad spectrum of cellular processes. Numerous critical cancer processes, such as tumor growth, metastasis, neovascularization, and chemotherapy resistance, have been shown to be significantly impacted by them. Protein kinases catalyze the transfer of a phosphate group from ATP to the hydroxy group of an amino acid residue. In cellular and molecular process, protein kinases are indispensable. As a result, they play a crucial part in the growth, dissemination, and survival of tumor cells in humans. Hence, this class of enzymes has drawn significant attention as a potential therapeutic target, with multiple kinase suppressors now receiving FDA approval for different cancer indications.
1. Introduction
2. Epidermal Growth Factor Receptor (EGFR) Inhibitors
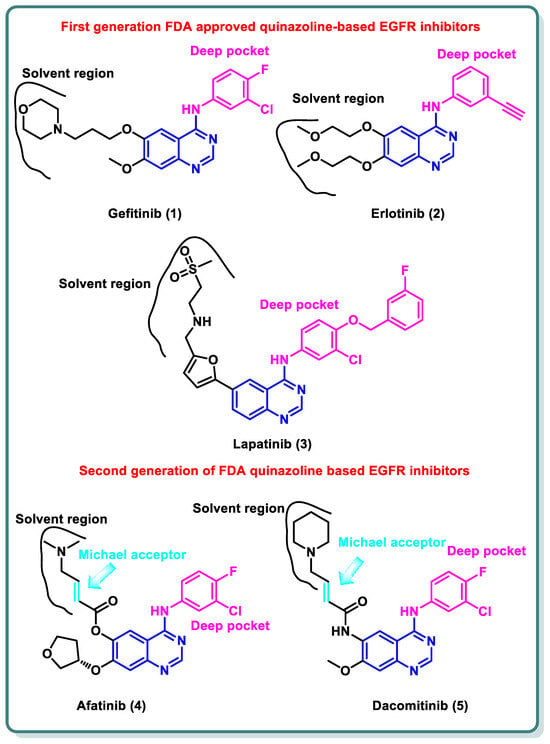
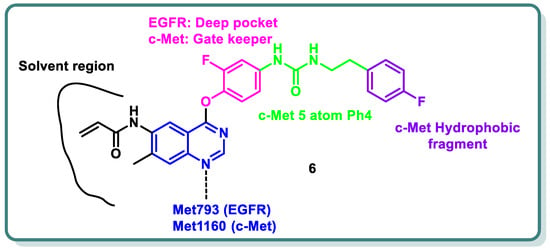
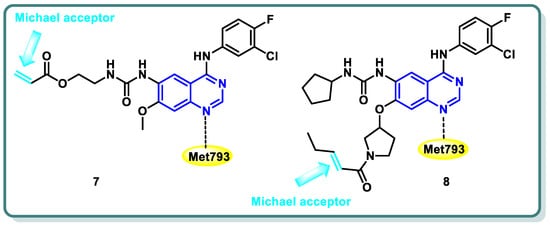
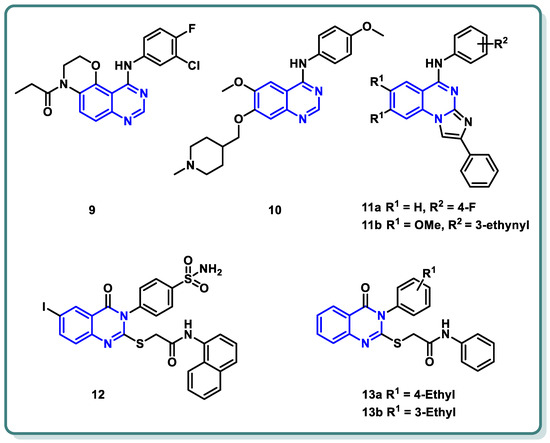
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190.
- Singh, D.; Vaccarella, S.; Gini, A.; De Paula Silva, N.; Steliarova-Foucher, E.; Bray, F. Global patterns of Hodgkin lymphoma incidence and mortality in 2020 and a prediction of the future burden in 2040. Int. J. Cancer 2022, 150, 1941–1947.
- Shenoy, G.P.; Pal, R.; Purwarga Matada, G.S.; Singh, E.; Raghavendra, N.M.; Dhiwar, P.S. Discoidin domain receptor inhibitors as anticancer agents: A systematic review on recent development of DDRs inhibitors, their resistance and structure activity relationship. Bioorg. Chem. 2023, 130, 106215.
- Giaquinto, A.N.; Miller, K.D.; Tossas, K.Y.; Winn, R.A.; Jemal, A.; Siegel, R.L. Cancer statistics for African American/Black People 2022. CA Cancer J. Clin. 2022, 72, 202–229.
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. CMLS 2008, 65, 1566–1584.
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748.
- Ranson, M. Epidermal growth factor receptor tyrosine kinase inhibitors. Br. J. Cancer 2004, 90, 2250–2255.
- Pal, R.; Teli, G.; Matada, G.S.P.; Dhiwar, P.S. Designing strategies, structural activity relationship and biological activity of recently developed nitrogen containing heterocyclic compounds as epidermal growth factor receptor tyrosinase inhibitors. J. Mol. Struct. 2023, 1291, 136021.
- Thomas, P.; Vincent, B.; George, C.; Joshua, J.M.; Pavithran, K.; Vijayan, M. A comparative study on erlotinib & gefitinib therapy in non-small cell lung carcinoma patients. Indian J. Med. Res. 2019, 150, 67–72.
- Xie, Y.; Ge, R.; Sang, D.; Luo, T.; Li, W.; Ji, X.; Yuan, P.; Wang, B. Real-world data of lapatinib and treatment after lapatinib in patients with previously treated HER2-positive metastatic breast cancer: A multicenter, retrospective study. Cancer Med. 2020, 9, 2981–2988.
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659.
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272.
- Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819.
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181.
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73.
- Wang, S.; Li, J. Second-generation EGFR and ErbB tyrosine kinase inhibitors as first-line treatments for non-small cell lung cancer. OncoTargets Ther. 2019, 12, 6535–6548.
- Li, H.S.; Wang, S.Z.; Xu, H.Y.; Yan, X.; Zhang, J.Y.; Lei, S.Y.; Li, T.; Hao, X.Z.; Zhang, T.; Yang, G.J.; et al. Afatinib and Dacomitinib Efficacy, Safety, Progression Patterns, and Resistance Mechanisms in Patients with Non-Small Cell Lung Cancer Carrying Uncommon EGFR Mutations: A Comparative Cohort Study in China (AFANDA Study). Cancers 2022, 14, 5307.
- Zhang, H.; Gan, W.; Fan, D.; Zheng, P.; Lv, Q.; Pan, Q.; Zhu, W. Novel quinazoline-based dual EGFR/c-Met inhibitors overcoming drug resistance for the treatment of NSCLC: Design, synthesis and anti-tumor activity. Bioorg. Chem. 2024, 142, 106938.
- Gan, W.; Wang, C.; Pan, Q.; Li, Y.; Guo, Y.; Fan, D.; Peng, Y.; Rao, Z.; Xu, S.; Zheng, P.; et al. Discovery of novel 4-arylamino-quinazoline derivatives as EGFR(L858R/T790M) inhibitors with the potential to inhibit the non-small cell lung cancers. Bioorg. Chem. 2022, 127, 105994.
- Qin, X.; Liu, P.; Li, Y.; Hu, L.; Liao, Y.; Cao, T.; Yang, L. Design, synthesis and biological evaluation of novel 3,4-dihydro-2H-oxazino quinazolin derivatives as EGFR-TKIs. Bioorg. Med. Chem. Lett. 2023, 80, 129104.
- Dou, D.; Wang, J.; Qiao, Y.; Wumaier, G.; Sha, W.; Li, W.; Mei, W.; Yang, T.; Zhang, C.; He, H.; et al. Discovery and optimization of 4-anilinoquinazoline derivatives spanning ATP binding site and allosteric site as effective EGFR-C797S inhibitors. Eur. J. Med. Chem. 2022, 244, 114856.
- Hasanvand, Z.; Oghabi Bakhshaiesh, T.; Peytam, F.; Firoozpour, L.; Hosseinzadeh, E.; Motahari, R.; Moghimi, S.; Nazeri, E.; Toolabi, M.; Momeni, F.; et al. Imidazoquinazolines as novel, potent EGFR-TK inhibitors: Design, synthesis, bioactivity evaluation, and in silico studies. Bioorg. Chem. 2023, 133, 106383.
- Ghorab, M.M.; Soliman, A.M.; El-Adl, K.; Hanafy, N.S. New quinazoline sulfonamide derivatives as potential anticancer agents: Identifying a promising hit with dual EGFR/VEGFR-2 inhibitory and radiosensitizing activity. Bioorg. Chem. 2023, 140, 106791.
- Ghorab, W.M.; El-Sebaey, S.A.; Ghorab, M.M. Design, synthesis and Molecular modeling study of certain EGFRinhibitors with a quinazolinone scaffold as anti-hepatocellular carcinoma and Radio-sensitizers. Bioorg. Chem. 2023, 131, 106310.


