Protein kinases are one of the largest enzyme families. By manipulating the location, activity, and functionality of many proteins via multisite phosphorylation, they regulate a broad spectrum of cellular processes. Numerous critical cancer processes, such as tumor growth, metastasis, neovascularization, and chemotherapy resistance, have been shown to be significantly impacted by them. Protein kinases catalyze the transfer of a phosphate group from ATP to the hydroxy group of an amino acid residue. In cellular and molecular process, protein kinases are indispensable. As a result, they play a crucial part in the growth, dissemination, and survival of tumor cells in humans. Hence, this class of enzymes has drawn significant attention as a potential therapeutic target, with multiple kinase suppressors now receiving FDA approval for different cancer indications.
1. Introduction
Cancer is widely recognized as a serious public health issue and is universally acknowledged as the second-greatest cause of death in the United States [
1]. According to the World Health Organization (WHO), the word “cancer” refers to a broad category of illnesses that can impact any area of the body [
2]. Other terms used to characterize cancer are neoplasms and malignant tumors [
3]. The fast growth of abnormal cells that eventually spread to other organs after initially residing in one area is one of the primary characteristics of cancer. This phenomenon is known as metastasis [
4]. Widespread metastases are the main reason behind cancer-related deaths [
4].
Recent WHO statistics indicate that 10 million deaths worldwide are expected to be related to cancer [
5]. The International Agency for Research on Cancer (IARC) estimates that one in five people will develop cancer in their lifetime. One of the most important public health issues of the twenty-first century is cancer prevention [
5]. Recent evidence suggests that effective primary prevention techniques could avoid at least 40% of cancer cases, and early tumor diagnosis could further reduce cancer-related death.
2. Epidermal Growth Factor Receptor (EGFR) Inhibitors
One member of the ErbB family of the transmembrane tyrosine kinase receptors is the epidermal growth factor receptor (EGFR) [
13]. It functions as a crucial mediator in cell proliferation, survival, adhesion, migration, and differentiation via downstream signaling transmission through auto-phosphorylation of several tyrosine residues following EGFR dimerization [
13]. This receptor is activated when ligands, such as EGF or TGFa, bind to the extracellular domain, which sets off a signal transduction cascade that leads to cell proliferation, metastasis, and resistance to apoptosis in many types of cancers. Numerous malignancies, including those of the breast, ovary, colon, and non-small cell lung cancers (NSCLC), are linked to overexpression of EGFR [
14].
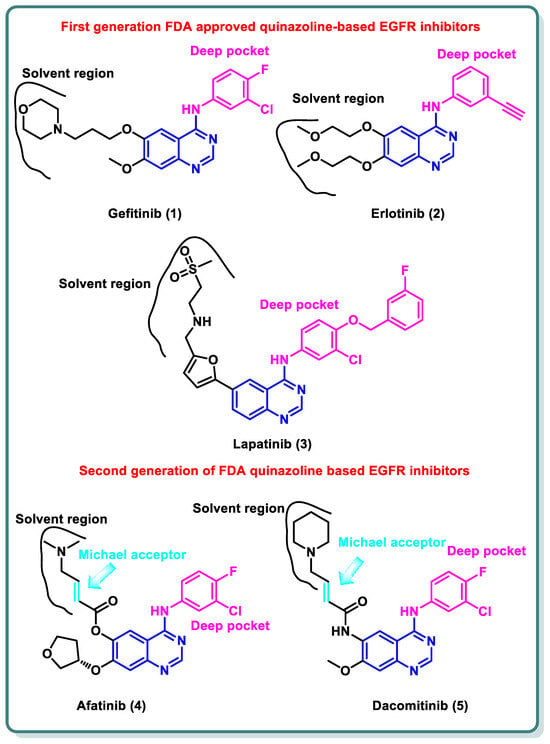
A variety of quinazolines have been discovered in recent years to inhibit EGFR [
15]. It is suggested that the 4-anilinoquinazoline scaffold interacts with ATP at the EGFR binding site [
16]. Gefitinib (
1) and erlotinib (
2), the first-generation EGFR inhibitors, have received FDA approval for the treatment of non-small cell lung cancer (NSCLC) [
17].
Additionally, lapatinib (
3) has received FDA approval for the management of certain cases of postmenopausal women’s breast cancer [
18]. Despite the advancements made, many non-small cell lung cancers (NSCLCs) develop EGFR binding site mutations that promote tumor formation and carcinogenesis. The protein databank incorporates the crystal structure of EGFR co-crystalized with gefitinib (
1), erlotinib (
2), and lapatinib (
3) [
19,
20]. These three inhibitors share the same binding mode to EGFR in which the quinazoline moiety occupies the ATP binding site and is stabilized by the ability of N1 to perform hydrogen bonding with the NH of Met793 in the hinge region, while N3 is involved in hydrogen bonding to Thr854 through a water molecule. The substituent at the 4-position is directed to the back of the ATP binding site and is involved in hydrophobic interactions with the protein, while the substitution at position 6 (and 7) is directed toward the solvent interface (
Figure 1) [
21].
Figure 1. Structures of first- and second-generation FDA-approved quinazoline-based EGFR inhibitors.
Despite the advancements made, many NSCLCs develop EGFR binding site mutations that promote tumor formation and carcinogenesis. About 40% of NSCLC patients have an activating mutation of L858R, which raises the affinity for ATP and reduces the affinity for first-generation TKIs, resulting in resistance [
22]. In contrast, about 50% of lung adenocarcinomas in patients who have become resistant to gefitinib (
1) and erlotinib (
2) exhibit the treatment resistance mutation T790M [
23]. It entails changing threonine 790 at the gate region to a bulkier methionine, which prevents inhibitors from binding to the EGFR binding site. Hence, various structural alterations were made to the previous drugs to provide a range of ATP-competitive irreversible EGFR inhibitors that were recognized as second-generation, irreversible EGFR inhibitors [
24]. Afatinib (
4) and dacomitinib (
5) occupy the ATP binding site of EGFR in the same manner as gefinitib (
1); however, they are classified as irreversible EGFR inhibitors, as they have dimethylaminocrotonamide and 4-piperidin-1-yl-but-2-enamide, moieties, respectively, at the sixth position. These are capable of forming a covalent bond with the Cys797 present EGFR’s ATP binding site by acting as a Michael acceptor (
Figure 1) [
25].
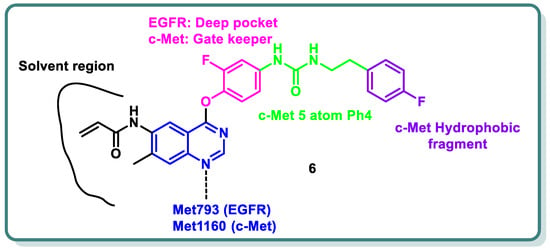
Additionally, different research groups have recently reported diverse scaffolds based on the quinazoline moiety as EGFR inhibitors beside other protein kinases. For example, in 2024, Han Zhang and his coworkers designed and synthesized novel 4-phenoxyquinazoline compounds as dual EGFR/c-Met suppressors to treat non-small cell lung cancer [
26] (
Figure 2). The most promising candidate is derivative
6, which inhibits EGFR, EGFR
L858R/T790M, and c-Met kinases at IC
50 values of 64.8, 305.4, and 137.4 nM, respectively. It has significant anti-proliferative activity (IC
50 = 2.27–3.35 µM), similar to that of afatinib, towards five types of cancer cell lines. Analysis of the cell cycle revealed that derivative
6 has the capacity to induce arrest at G2/M. In vivo findings in xenograft models demonstrated that derivative
6 might both cause apoptosis and suppress tumor development. Altogether, the results showed that derivative
6 is a novel compound with dual inhibitory activity against both EGFR and c-Met, and it can be considered a potential therapeutic medication to treat a variety of tumors (
Figure 2) [
26].
Figure 2. Design strategy of quinazoline derivative 6 as a dual EGFR and cMet inhibitor.
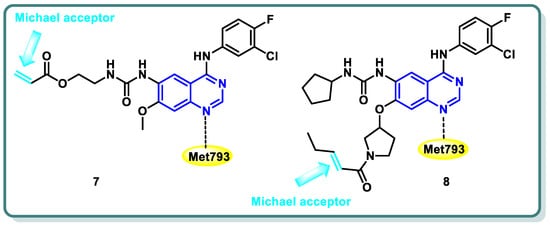
Diverse series of quinazolines were designed as EGFR
L858R/T790M inhibitors in 2022 by Wenhui Gan and coworkers [
27]. Urea and thiourea moieties were incorporated in one series (compound
7 is a representative example,
Figure 3), whereas a Michael receptor active warhead was introduced in another series (compound
8 is a representative example,
Figure 3). Most of the designed and synthesized candidates demonstrated potent anti-proliferative impacts against different cell lines, including A549 and H1975 cancer cells. Additionally, they demonstrated moderate to outstanding kinase inhibitory efficacy against EGFR
WT and EGFR
L858R/T790M. Urea derivative
7 revealed the most promising EGFR
WT and EGFR
L858R/T790M inhibitory activities (IC
50 = 0.8 and 2.7 nM, respectively). Furthermore, both derivatives
7 and
8 strongly caused apoptosis of A549 cells. A cell cycle analysis showed that arrest of the cell cycle of A549 cells occurred at the S phase in derivative
7 and the G1 phase in derivative
8 [
27] (
Figure 3).
Figure 3. Structures of quinazoline derivatives 7 and 8.
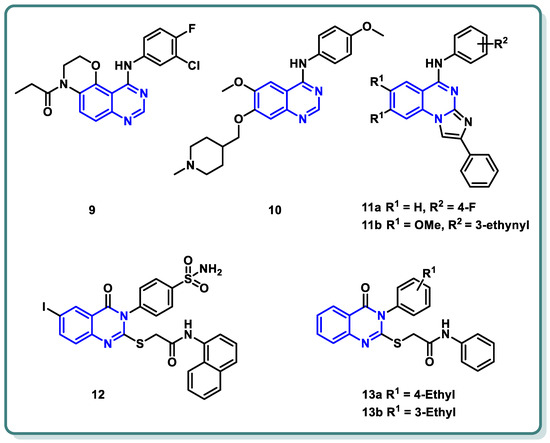
Moreover, a new family of 3,4-dihydro-2
H-[1,4]oxazino [2,3-
f]quinazoline derivatives that target EGFR was described by Qin et al. (
Figure 4) [
28]. With an IC
50 ≤ 937.7 nM, the synthesized candidates showed strong EGFR inhibitory efficacy. Compound
9 (
Figure 4) exhibited strong anti-proliferative properties against NCI-H1563 and H1975 cancer cell lines (IC
50 = 25.69 nM). Furthermore, it produced an in vitro safety profile against the normal cell line, resembling 16HBE cells [
28].
Figure 4. Chemical structures of various substituted quinazolines-based compounds 9–13 as EGFR inhibitors.
A series of 4-anilinoquinazoline analogues that are expected to be accommodated in the hinge region and allosteric pocket of EGFR
C797S was designed by Dou Dou [
29]. When screening the designed and synthesized quinazolines regarding their inhibitory activity against EGFR
C797S, derivative
10 (
Figure 4) displayed a potent activity, with an IC
50 = 0.128 µM. A potential anti-proliferative impact against BaF3-EGFR
L858R/T790M/C797S and BaF3-EGFR
19del/T790M/C797S was recorded for derivative
10 (IC
50 = 0.75 and 0.09 μM, respectively). Additionally, derivative
10 demonstrated dose-dependent inhibition of EGFR and its downstream signaling cascades in BaF3-EGFR
19del/T790M/C797S cells. Interestingly, quinazoline
10 (
Figure 4) demonstrated in vivo inhibition of tumor growth in a BaF3-EGFR
19del/T790M/C797S xenograft model (30 mg/kg, TGI = 67.95%) [
29].
In 2023, a class of imidazo [1,2-
a]quinazolines was presented by Hasenvand et al. as containing potent EGFR inhibitors [
30]. When compared to erlotinib, the reference drug, imidazoquinazolines,
11a,
b (
Figure 4) revealed significant anticancer activity against PC3, HepG2, HeLa, and MDA-MB-231, with IC
50 values in the micro-molar range. Additional analyses showed that they could cause cell growth inhibition at the G0 phase of the cell cycle and induce apoptotic cell death. Two derivatives,
11a,
b were shown to have suppression activity and selectivity toward EGFR (as seen by their respective EGFR-IC
50 = 82.0 µM and 12.3 µM, respectively). Furthermore,
11a,
b were shown via Western blot analysis to decrease extracellular signal-regulated kinase (ERK1/2) as well as EGFR phosphorylation. The degree of B-Actin phosphorylation, however, remained unchanged [
30] (
Figure 4).
In addition, Ghorab et al. introduced a series of novel quinazoline sulfonamide derivatives in 2023 [
31]. Using the MTT assay, the compounds’ growth inhibitory activities against four cancer cell lines, namely HepG2, MCF-7, HCT116, and A549, were assessed. Based on the obtained findings, the compounds showing the most significant potencies were examined as EGFR
T790M and VEGFR-2 inhibitors. With an IC
50 = 0.0977 µM against MCF-7, the quinazoline sulfonamide derivative
12 (
Figure 4) exhibited the most noticeable cytotoxic effect and the most inhibitory activity against EGFR
T790M and VEGFR-2 (IC
50 = 0.0728 and 0.0523 µM, respectively). In MCF-7 cells, compound
12 induced apoptosis and cell cycle stopping at the G2/M phase. Following an 8 Gy gamma irradiation dose, the radiosensitizing evaluation of compound
12 demonstrated its noteworthy capacity to sensitize the cancer cells to the impact of radiation. According to molecular docking simulation studies, compound
12 was predicted to occupy the VEGF and EGF receptors’ ATP-binding site and block their functions [
31] (
Figure 4).
The same research group designed and synthesized a class of
N-substituted-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamides [
32]. Some of the synthesized candidates demonstrated potent cytotoxic activity on a HepG2 cell line with an IC
50 reaching 1.11 µM. The quinazolines
13a and
13b inhibited EGFR (IC
50 = 73.23 and 58.26 µM, respectively) in reference to erlotinib (IC
50 = 9.79 µM) [
32] (
Figure 4).
This entry is adapted from the peer-reviewed paper 10.3390/molecules29040875