Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesco Nappi | -- | 4461 | 2024-03-24 07:48:03 | | | |
| 2 | Camila Xu | + 116 word(s) | 4577 | 2024-03-26 03:07:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nappi, F. Current Knowledge of Enterococcal Endocarditis. Encyclopedia. Available online: https://encyclopedia.pub/entry/56465 (accessed on 25 July 2026).
Nappi F. Current Knowledge of Enterococcal Endocarditis. Encyclopedia. Available at: https://encyclopedia.pub/entry/56465. Accessed July 25, 2026.
Nappi, Francesco. "Current Knowledge of Enterococcal Endocarditis" Encyclopedia, https://encyclopedia.pub/entry/56465 (accessed July 25, 2026).
Nappi, F. (2024, March 24). Current Knowledge of Enterococcal Endocarditis. In Encyclopedia. https://encyclopedia.pub/entry/56465
Nappi, Francesco. "Current Knowledge of Enterococcal Endocarditis." Encyclopedia. Web. 24 March, 2024.
Copy Citation
Enterococci are a unique type of bacteria due to their ability to withstand a broad range of different environmental parameters such as pH, temperature, salinity, bile acids, and so on. They are resistant to many antibiotic compounds and have the flexibility to flourish as both common commensal and opportunistic pathogens in a broad range of clinical settings.
Enterococcus faecalis
vegetation
infective endocarditis
biofilm
cardiac endovascular infection
1. Introduction
Enterococci are a unique type of bacteria due to their ability to withstand a broad range of different environmental parameters such as pH, temperature, salinity, bile acids, and so on. They are resistant to many antibiotic compounds and have the flexibility to flourish as both common commensal and opportunistic pathogens in a broad range of clinical settings [1][2][3][4][5][6]. Enterococci commonly live in the body and can cause chronic endocarditis, especially Enterococcus faecalis [5][7][8][9][10]. They account for approximately 10% of valvular endocarditis cases, with E. faecalis being the main causative agent [6][8][9][10][11][12][13].
To improve patient outcomes, it is important to accurately diagnose and treat enterococcal infections. During colonisation of the murine gastrointestinal (GI) tract, E. faecalis has been shown to form and develop bacterial biofilms. These biofilms consist of bacteria attached to a host surface and surrounded by a bacterially derived extracellular matrix (ECM) [13][14]. In animal models of enterococcal catheter-associated urinary tract infections and endocarditis, E. faecalis has been identified as a significant pathogenic factor [15][16][17][18][19][20][21][22]. This finding was first reported in 2007 [22]. Colonisation results in the formation of a defensive bacterial biofilm on the native or engineered tissue: biofilm formation often results in markedly enhanced levels of resilience to antimicrobial agents [23][24][25].
Bacteria colonise a prestaged, abacterial collection of host factors according to the classical or canonical model of bacterial endocarditis. The model proposes a two-step process: First, platelets, components of the coagulation chain including fibrinogen, thrombin, etc., and other host factors are deposited in reaction to an initial injury, thereby creating a “sterile vegetation”. Bacteria already circulating in the bloodstream then populate this aberrant site, establishing a largely quiescent infection nidus [11]. Likewise, enterococcal infection is a significant cause of dysfunction in allogeneic tissue used as a biological valve replacement for patients who have received an allograft, whether for endocarditis or non-endocarditis of the aortic valve [12][26].
Barnes et al. [14] have previously reported that E. faecalis directly engages and colonises the surface of the intestinal epithelium, producing distinct biofilm microcolonies across the gastrointestinal tract in a germ-free mouse model of infection. In a rabbit model of cardiac endovascular infection, a comparable pattern of colonisation of the native host surface is also observed [13]. These observations suggest that the adhesion of enterococci to the cardiac endothelium has a similar role in the development of pathogenic endocarditis as it does in non-pathogenic intestinal epithelial colonisation. This is supported by the absence of significant systemic host responses to this colonisation over several weeks and the ability of E. faecalis to adhere to intact endothelia.
2. History
Originally described in the early 20th century and named Streptococcus faecalis before being placed in the genus Enterococcus in 1984, Enterococcus faecalis has been known to cause endocarditis since the seminal paper published by Andrewes and Horder in 1906 [27].
As previously mentioned, the conventional paradigm for bacterial colonisation of the heart involves an abiotic accumulation of host factors. This is usually accompanied by an endovascular injury. Nevertheless, it is noteworthy that numerous papers in the earlier literature (prior to 1975) reported that enterococcal endocarditis appeared to arise in a substantial proportion of individuals without obvious prior gross endothelial damage or structural cardiac defects [28][29]. As is frequently the case in earlier literature, the exact determination of the particular bacterial strain can be challenging. Several animal model models, notably pigs [30] and rabbits [31], have also described these clinical findings.
During the 1970s and 1980s, the medical community focused on enterococci because of their high level of intrinsic and transmissible antibiotic resistance in comparison to pathogenic streptococci. It is worth noting that until the 1980s, enterococci were phylogenetically classified as members of the genus Streptococcus [32].
The interest of the medical and health care community in enterococci during the 1970s and 1980s was largely driven by the relatively high level of inherent and transmissible antibiotic resistance of these bacteria compared to the pathogenic streptococci routinely found in the population. It is noteworthy to mention that enterococci were classified phylogenetically as belonging to the species Streptococcus right up to the 1980s [32]. During this time, genetic and molecular studies of both plasmids and trans-spliced genetic material provided an important experimental basis for future genomic approaches to enterococcal virulence [33][34]. Yet, the global clinical frequency of clinically ascertained enterococcal infections continued to be low throughout much of this time frame, although it is unclear whether this represents a real incidence rate or merely a reflection of a more restricted diagnostic landscape.
In the 1980s, the widespread use of oral prophylaxis with cephalosporins led to the emergence of enterococci (mainly E. faecalis) as the most important hospital pathogens. Certain genotypes were able to achieve epidemic dissemination, both nationally and internationally [35][36][37][38][39][40][41]. Starting in the 1990s, systematic attempts to determine crucial genetic factors of virulence in nosocomial and other opportunistic enterococcal infections were intensified as a result of these clinical developments. Pioneering studies in this field aimed to identify enterococcal antigens that triggered an antibody response in patients with infections [42][43][44][45][46][47][48][49][50]. In early studies, most of the prominent antigens discovered were surface-exposed antigens of the enterococcal cell coat (Ebp, Ace, Epa). Subsequent studies using in vitro assays and animal models, including experimental endocarditis, have identified critical roles for these constituents in host adherence and virulence [42][43][44][45][46][47][48][49][50]. In addition to the factors mentioned above, which are genetically determined, there is also evidence that plasmid-encoded surface adhesins, such as Aggregation Substance, play a role [51][52][53][54][55][56][57].
Enterococci have become increasingly significant in healthcare-associated infections over the last two decades. This trend is likely to be driven by a number of factors. Among them are increased access to diagnostics, an increasingly elderly population, greater invasiveness of medical interventions, and the continued emergence of antimicrobial resistance [58][59][60][61][62][63]. During this time period, the number of studies in the general area of bacterial biofilms increased markedly [64][65][66][67][68][69][70][71][72]. Additionally, the full genome sequence for E. faecalis V583 was published [73][74][75]. In 2003, Bourgogne et al. [76] identified OG1RF, and since then, several other strains have been extensively studied [77] using enhanced genetic research tools to investigate E. faecalis infection [78][79][80][81][82]. Barnes et al. [13][14] conducted a thorough study on transposon mutagenesis and recombinase-based in vivo expression technology (RIVET) genetic screens. The results were non-overlapping but mutually supportive, identifying several factors involved in multiple in vitro biofilm production in the chromosome of strain OG1RF. These findings were previously reported by Kristich et al. [80] and Ballering et al. [81]. When the same RIVET library was tested in a rabbit model of subcutaneously implanted foreign body infection, 28 genes identified in these in vitro tests (two from the transposon screen, 26 from the RIVET screen) were also found to have promoters [19]. However, only two genes (ahrC and eep) were considered to play a significant role in endocarditis pathogenesis when ten strains with mutations in biofilm-associated genes from these candidate genes were tested for in vivo virulence impairment in a rabbit model of infective endocarditis [19][83]. Leuck et al. [84] found that E. faecalis clinical strains that were classed as poor biofilm producers in a standard in vitro microtiter dish assay colonised porcine heart valves in an ex vivo assay just as well as strong biofilm-forming clinical strains, supporting the conclusion that in vitro biofilm phenotypes do not closely predict infective endocarditis.
Madsen et al. conducted a systematic literature review that summarised nine virulence factors of E. faecalis infective endocarditis [17]. This information is highly useful for readers. The virulence factors listed therein comprise the aggregation substance, cell wall glycolipids, the Ebp pili proteins, haemolysin, the stress protein gls24, the secreted protease GelE, the membrane metalloprotease Eep, and the adhesins Ace and EfbA [17][85]. The transcriptional regulator AhrC is the tenth virulence factor of E. faecalis endocarditis. It affects the expression of the ace and ebp genes, as reported by Frank et al. [19] and Manias and Dunny [54][86][87] (Figure 1 [88][89][90][91]).
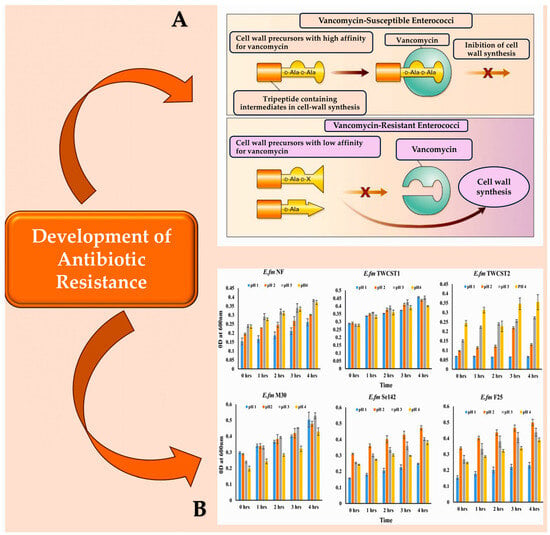
Figure 1. (A): Vancomycin-susceptible enterococci synthesise cell wall precursors that bind vancomycin with high affinity. These precursors end in D-Ala-D-Ala and are translocated from the cytoplasm to the cell surface where, once bound, they cannot participate in cell wall synthesis. In the presence of an inducer like vancomycin, vancomycin-resistant enterococci produce intermediates with different end groups (D-Ala-D-Lac, D-Ala, or D-Ala-D-Ser), which have a low affinity for vancomycin and can therefore be used for cell wall synthesis. (B): The resistance of the chosen enterococcal strains to artificial gastric juice, containing pepsin and acidified to pH 1.0 to pH 4.0, was tested by incubating them for 4 h at 37 °C. This mimics the transient time food spends in the stomach. Abbreviations: ‘LA’ denotes either alanyl or alanine, while ‘X lactate’ is used for VanA, VanB, and VanD types of resistance, and ‘serine’ is used for VanC and VanE types [88][89][90][91].
The genetic drivers involved in E. faecalis biofilm formation are shown in Table 1 [92]. Table 1 Shows the genetic determinants that are involved in the formation of E. faecalis biofilm [47][92].
Table 1. Genetic determinants that are involved in the formation of E. faecalis biofilm.
| Gene/Locus | Protein/Function | Year of Publication |
|---|---|---|
| srtC | Sortase C/an enzyme that anchors surface proteins to the cell wall | 2006 [47] |
| atn | Autolysin | 2004 [92] |
| salB | Secretory antigen-like B/cell-shape determinant | 2004 [92] |
| bee | Biofilm enhancer in Enterococcus/a putative cell wall-anchored protein | 2006 [93] |
| salA | Secretory antigen-like A | 2004 [92] |
| bop | Biofilm on plastic surface/a putative sugar-binding transcriptional regulator | 2004 [94] |
| gelE | Secretory metalloprotease gelatinase E | 2004 [8][92][95] |
| dltA | D-alanine lipoteichoic acid/D-alanine-D-alanyl carrier protein ligase | 2006 [96] |
| ebpA, ebpB, ebpC | Endocarditis and biofilm-associated pili | 2006 [47] |
| ebpR | Transcriptional regulator of ebpABC | 2007 [97] |
| epa (orfde4) | Enterococcal polysaccharide antigen/a putative glycosyltransferase involved in polysaccharide synthesis |
2004 [92] |
| esp | Enterococcal surface protein | 2001, 2004 [91][98] |
| etaR | Enterococcal two-component system regulator | 2004 [92] |
| fsrA, fsrB, fsrC | E. faecalis regulator/two-component quorum-sensing signal transduction system, regulates the expression of gelatinase and serine protease. | 2004 [8][92][95] |
3. Causes of E. faecalis Bacteraemia
Bacteraemia is evidently required for endothelial bacterial colonisation of the endothelium and the development of IE. In cases of acute bacteriaemia, the initial source of infection is often identifiable. This is due to the short period of time between the spread of bacteraemia and the onset of IE. Chronic endocarditis, which is similar to the classic enterococcal endocarditis, is often much more ambiguous [94][99]. A variety of causes have been proposed, varying from colonisation of the oral cavity in endodontic disease to translocation of commensal enterococci in the gastrointestinal tract [95][96]. Enterococcus is the second leading cause of hospital-acquired bacteraemia, due in part to its ability to thrive in challenging environments. Contamination of environmental surfaces in healthcare settings can cause exogenous infection, leading to direct seeding of the vasculature through catheterisation or contamination of implantable medical devices. Indirect infection can also occur through colonisation of the urinary or gastrointestinal tracts. Endogenous infections can also result from translocation through the epithelium of the GI tract [95][96][97][98][100][101] (Figure 2).
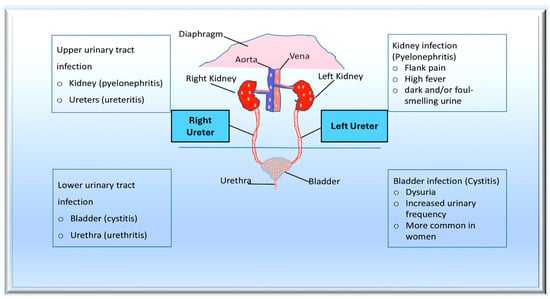
Figure 2. The pathogenesis of urinary tract infections (UTIs) begins with the colonisation of uropathogens in the urethra, followed by the bladder, facilitated by specific adhesins. Causative bacteria proliferate and form biofilms if they are not eliminated by the immune system. Pathogens can ascend from the lower urinary tract to the kidney, leading to bacteraemia. Uropathogens can bind to the catheter and multiply within the biofilm in the case of complicated UTIs. The infection may progress to pyelonephritis and bacteraemia if left untreated. From Mancuso et al. [97].
This process is facilitated by conventional antibiotic regimens, which can drastically increase the number of enterococci in the intestinal flora [102][103]. More than three decades ago, Wells et al. [104] experimentally demonstrated translocation of E. faecalis across the epithelial barrier of the GI system and subsequent penetration into the circulation in a mouse model. More advanced work has followed, including detection of invasion-defective E. faecalis mutant strains in a T84 cell culture model [105][106][107] and high-resolution imaging of the process with complementary findings on intracellular migration [108]. Despite the long-standing belief that oral enterococci are a likely source for endocarditis, cohort evidence has shown that oral infections are not a common factor in IE, despite the fact that enterococci are also commonly found in the oral cavity and are a leading etiology of endodontic disease [108]. For instance, only 1.6% of enterococcal cases could be attributed to oral routes of transmission versus 6.7% of non-enterococcal cases in a recent large Spanish cohort study comparing enterococcal IE (516 patients) and non-enterococcal IE cases (3308 patients) [59].
Severe physiological challenge, in combination with the possibility of organism-specific translocation, may result in enough GI barrier breakdown to permit bacterial penetration via systemic host immunosuppression [109][110][111][112]. It is unclear whether enterococcal translocation is a result of host immunosuppression or if enterococci themselves are immunomodulatory and can initiate the suppressive response [110]. In a mouse model, common antibiotics at clinically relevant doses can cause GI barrier dysfunction and bacterial translocation, in some cases after a single dose. Again, E. faecalis is a key player [113][114][115].
Brown et al. [116] have recently reported the discovery of cardiac microlesions during severe bacteraemia caused by E. faecalis infection in mice. These microinjuries are similar to those caused by Streptococcus pneumoniae during invasive pneumococcal disease. However, E. faecalis does not encode the virulence determinants involved in pneumococcal microinjury formation. The study discovered that the protein DsbA, which forms disulphide bonds, is essential for E. faecalis virulence in a C. elegans model and for the efficient formation of cardiac microlesions. Additionally, E. faecalis facilitated necroptotic cell death of cardiomyocytes at sites of microlesion formation. Unlike the wild-type strain, which suppressed the immune response, loss of DsbA resulted in an increase in pro-inflammatory cytokines. Furthermore, E. faecalis was able to induce microlesions in the heart. This study has identified the features of both the bacterium and the host response that are involved in this process.
Although there is only a paucity of clinical evidence to date, there is also some emerging data on an association between enterococcal endocarditis events and cryptic colorectal cancers [117][118][119]. It is uncertain whether there is a significant association between these clinical conditions, as seen in most cases of Streptococcus gallolyticus subsp. gallolyticus endocarditis, previously associated with Streptococcus bovis biotype I [120][121][122][123][124]. Stanley et al. [125] found that a murine model of ischemic-reperfusion stroke showed bacteraemia caused by a specific group of commensal bacterial strains, with enterococci being the most prevalent.
3.1. Induced Enterococcal Colonisation Involves Cell Surface Mechanisms—Ultra-Large von Willebrand Factor and Sortase Are Key Players in This Process
The accepted developmental pathway for bacterial endocarditis includes the primary production of a host-derived thrombus, with subsequent processes promoting colonisation of the thrombus by bloodstream bacteria. However, there are multiple instances where direct colonisation of host epithelial surfaces has been reported and in practice, this mode of adhesion may be more prevalent than is currently recognised. S. aureus is one of the most studied of those bacterial pathogens that have been demonstrated to directly adhere to the endothelium, at least under some circumstances. S. aureus expresses three fundamental molecules on its surface: fibronectin-binding protein A (FnBPA) and B (FnBPB), as well as clumping factor A (ClfA). These molecules promote bacterial adherence and identify the cultured human endothelial cells (ECs) that interact with Gram-positive cocci. Three recent reports have investigated the adherence of Gram-positive cocci to endothelial cells (ECs) and have highlighted the fundamental importance of these molecules in IE [126][127][128].
Pappelbaum et al. [129] showed that Staphylococcus aureus adhesion to healthy endothelial cells is associated with elevated levels of ultra-large von Willebrand factor, a host cofactor that deserves in-depth analysis due to its peculiarities of action. Bacterial proteins, such as ClfA and FnBPA, help S. aureus stick to EC surface molecules. This is also done by subendothelial matrix proteins, like fibrinogen, fibrin, fibronectin, and von Willebrand factor (vWF). In the setting of the undamaged endothelium, evidence suggests that ultra-large von Willebrand factors (ULVWFs) significantly facilitate the initial pathogenic phase of S. aureus-induced endocarditis. When activated human endothelial cells were perfused with fluorescent bacteria under high-shear-rate conditions, 95% of the S. aureus attached to ultra-large von Willebrand factor (ULVWF) [129]. Flow experiments using VWF deletion mutants and heparin indicated that the A-type domains of VWF contribute to bacterial binding. The role of wall teichoic acid, but not staphylococcal protein A, was suggested by analysis of several bacterial deletion mutants. ULVWF-mediated bacterial adherence significantly increased with the presence of inactivated platelets and serum. ADAMTS13, a thrombospondin 13 disintegrin and metalloproteinase, reduced bacterial binding and shortened the length of ULVWF in a dose-dependent manner, but even at physiological levels of ADAMTS13, individual cocci remained bound by ULVWF. To further demonstrate the role of VWF in vivo, wild-type mice were compared with VWF knockout mice. Using the dorsal skinfold chamber model and intravital microscopy, fluorescent bacteria binding was observed in tumour necrosis factor-α-stimulated tissue. VWF knockout mice had fewer bacteria in their postcapillary and collecting venules compared to wild-type mice. Using heparin and ADAMTS13 can reduce ULVWF formation and may provide a novel therapeutic option to prevent IE [129].
Research has been conducted on the cell biology of NETosis in the context of infection [130]. The enzyme PAD4, which stands for protein arginine deiminase 4, plays a crucial role in this process. PAD4 is the only member of the PAD family that possesses a nuclear localisation signal [131][132][133][134]. Furthermore, it is believed that PAD4 has particular targets within the cytoplasm that affect the cell biology of NETosis and the composition of the neutrophil inflammasome. During an infection, functional cytoplasts (enucleated cells) capable of supporting phagocytosis can be identified. In blood vessels, NETs act as a platform for platelet adhesion and initiation of coagulation, similar to VWF [132][133][135][136]. Active PAD4, which is released in conjunction with NETs, also facilitates the citrullination of ADAMTS13. This impedes VWF scission and allows platelet aggregates to remain close to the vessel wall in the presence of PAD4 [137][138]. Recent studies have linked NETosis and the increase in NET-associated tissue factor (TF) to systemic inflammation and IL-1β levels, indicating a common regulatory pathway [139]. Additionally, TF secretion from activated macrophages and monocytes is stimulated by the activation of both canonical and non-canonical inflammasomes, as demonstrated by recent research [140][141] (Figure 3).
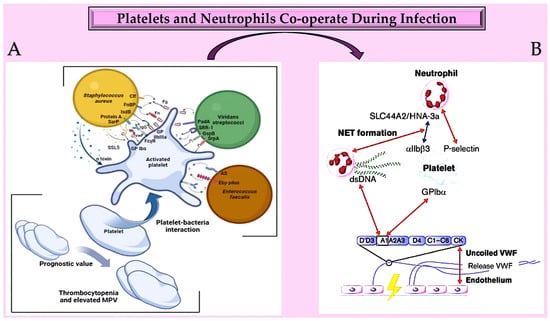
Figure 3. (A) Platelets play a crucial role in infective endocarditis. The interactions between platelets and the bacterial species involved in IE are complex and involve numerous ligand–receptor pairs. Changes in platelet parameters have predictive value. Additionally, von Willebrand factor is also involved. (B): The diagram illustrates the interactions between von Willebrand factor (VWF) and neutrophils at an infection site. It provides insights into the relationships between the A1 domain of VWF multimers, platelets, neutrophils, and NETs under conditions of high and low shear flow (indicated by red and blue arrows, respectively). Abbreviations: AS, aggregation substance; Clf, clumping factor; Ebp, endocarditis- and biofilm-associated pili; FcγR, crystallisable fragment gamma receptor; Fg, ds, double strand; fibrinogen; Fn, fibronectin; FnBP, fibronectin-binding protein; GP, glycoprotei; GspB, Streptococcus gordonii surface platelet B; IgG, immunoglobulin G; IsdB, iron-responsive surface determinant B; PadA, platelet adherence protein A; Sar P, staphylococcal accessory regulator protein; SrpA, serine-rich glycoprotein A; SRR-1, serine-rich repeat glycoprotein 1; SSL5, staphylococcal superantigen-like 5; VWF, von Willebrand factor [126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142].
The role of vWbp and the sortase-assembled pilus family emerged during the analysis of adhesion mechanisms in Gram-positive cocci infections. Claes et al. [126] discovered that the interaction between vWbp and surface proteins of S. aureus reduces bacterial adhesion to VWF and vascular endothelium under shear stress. Mutants deficient in Sortase A (SrtA) and SrtA-surface proteins, as well as Lactococcus lactis-transmitting single staphylo-surface proteins, have been employed. S. aureus attaches to the endothelium via vWF. The VWF-binding protein (vWbp) facilitates adhesion under shear stress. The vWbp interacts with vWF to complete the adhesion process. It is suggested that the synergistic action of Sortase, a ClfA-dependent surface protein, plays a role in this process.
Similarly, Enterococcus faecalis is an opportunistic bacterium that causes various hospital-acquired infections, including catheter-associated urinary tract infections. It may contribute to virulence and the development of infective endocarditis. In a mouse model of E. faecalis-ascending urinary tract infections, the role of the endocarditis- and biofilm-associated pilus (Ebp), a member of the sortase-assembled pilus family, was demonstrated. The Ebp pilus consists of the major EbpC shaft subunit and the minor subunits EbpA and EbpB. In experimental catheter-associated urinary tract infections, the EbpABC(-) strain, a non-piliated pilus knockout mutant, was significantly less virulent than its isogenic parent OG1RF. In contrast, the EbpC(-) strain, which is a mutant with a deleted nonpiliated ebpC gene, exhibited similar behaviour to OG1RF in vivo because it expressed EbpA and EbpB. Deletion of either the minor pilin gene ebpA or ebpB disrupted pilus biogenesis and resulted in defects in experimental catheter-associated urinary tract infections. The Ebp pilus has been identified as a virulence factor in E. faecalis catheter-associated urinary tract infections. Its in vivo function depends on a metal ion-dependent adhesion site motif that is predicted in EbpA’s von Willebrand factor A domain. Understanding the molecular basis of this common protein domain among the tip subunits of sortase-assembled pili is important in preventing and treating catheter-associated urinary tract infections caused by Enterococcus faecalis. The Ebp pilus of E. faecalis and its subunits are crucial to the virulence of enterococcal infections in a mouse model of catheter-associated urinary tract infections. The metal ion-dependent adhesion site motif in EbpA is crucial for Ebp function in vivo. This discovery has implications for the molecular basis of virulence in E. faecalis catheter-associated urinary tract infections, as well as other infections caused by enterococci and other Gram-positive pathogens. The metal ion-dependent adhesion sitemotif is also present in other sortase-assembled pili [126].
3.2. The Role of the Endocardium and Enterococcal Pathoadaptation
The endothelium is a specialised type of epithelium. This concept offers an intriguing explanation. Several studies have confirmed that the endocardium is indeed a modified endothelium [143][144][145][146][147], although there has been some uncertainty about the specifics of endocardial development. E. faecalis can directly colonise different host epithelial surfaces in a variety of animal experimental models. In a germ-free mouse model, Barnes et al. [14] demonstrated that E. faecalis can successfully colonise the surface of the intact, normal intestinal epithelium directly. Barnes et al. [13] have recently suggested that enterococcal coverage of endocardial and endovascular surfaces is possible without the need for host tissue destruction or even restricted surgical intervention, using a rabbit model of endocarditis.
Endocarditis caused by E. faecalis is a serious clinical manifestation, commonly acquired in a community setting. Understanding the extrinsic pathogenesis at the valve level is a priority. Infective endocarditis is a complex disease with many host and microbial components contributing to the formation of bacterial biofilm-like vegetations on the aortic valve and adjacent areas of the heart. Thurlow et al. [21] reported further evidence supporting a non-valvular role in early endocardial colonisation. In their model, even after the inflamed valve was harvested, cardiac tissue homogenates still showed greatly elevated bacterial loads.
In a rabbit model of enterococcal endocarditis, the pathogenic capacity of vancomycin-resistant E. faecalis V583 and three isogenic protease mutants (ΔgelE, ΔsprE, and ΔgelE ΔsprE mutants) were compared [148]. Compared to V583 or the SprE(-) mutant, the bacterial load in the heart of the GelE(-) mutants (ΔgelE and ΔgelE ΔsprE mutants) was considerably reduced. A marked deposition of the fibrinous matrix layer and increased chemotaxis of inflammatory cells was also observed on aortic valves infected with GelE(-) mutants (ΔgelE and ΔgelE ΔsprE mutants). This suggests a role for proteolytic modulation of the immune response to E. faecalis. Furthermore, it was observed that GelE can degrade the anaphylatoxin complement C5a and that this proteolysis leads to reduced neutrophil recruitment in vitro, supporting a role for proteolytic modulation of the immune response to E. faecalis. In vivo, GelE-producing strains were observed to cause a decrease in heterophil migration at infected tissue sites, while SprE-producing strains did not show this effect. These results indicate that of the two enterococcal proteases, GelE is the most important in mediating the pathogenesis of endocarditis. Perez et al. published an important study in which the gene encoding gelatinase (gelE) was found to be under the control of the Fsr quorum sensing system, whose encoding genes (fsrA, fsrB, fsrC, and fsrD) are situated immediately upstream of gelE. Biofilm formation was prevented and gelatinase activity was suppressed in a derived mutant of E. faecalis V583 when a DNA fragment was integrated into the fsr locus. Sequence analysis revealed the presence of IS256 integrated into the fsrC gene at nucleotide position 321. It is worth noting that IS256 is also linked to biofilm formation in Staphylococcus epidermidis and Staphylococcus aureus [148] (Figure 4 [149]).
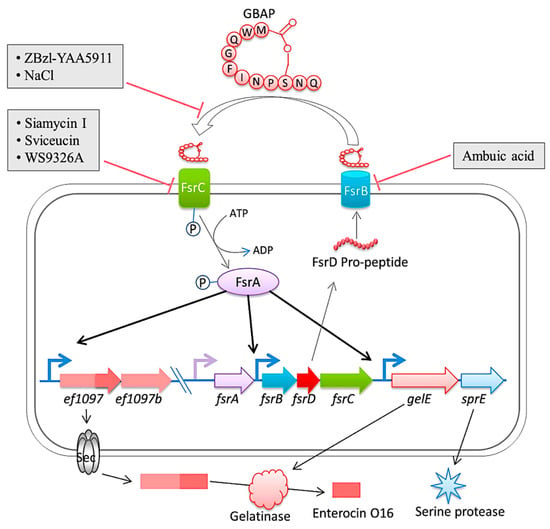
Figure 4. The diagram is a representation of the Fsr quorum sensing system and its regulation in E. faecalis. The system involves the export and processing of the FsrD propeptide (encoded by fsrD) to produce the small lactone gelatinase biosynthesis activating pheromone (GBAP) via FsrB. FsrC is part of a two-component regulatory system that reacts to extracellular GBAP by phosphorylating the intracellular response regulator FsrA, which in turn induces the expression of ef1097, ef1097b, the fsr locus, gelE (encoding a gelatinase) and sprE (encoding a serine protease). The pre-protein encoded by ef1097 is cleaved and transported via the Sec-dependent pathway and the precursor is further truncated by the gelatinase to form enterocin O16. The interaction of GBAP with FsrC is inhibited by ZBzl-YAA5911 in a competitive manner, whereas NaCl inhibits it in a concentration-dependent manner. The inhibition of FsrB activity is due to the presence of ambuic acid. Siamycin I, sviceucin, and WS9326A inhibited the phosphorylation of FsrC. From Ali et al. [149].
Enterococcal pathoadaptation to the endocardium is believed to be facilitated by the IS256 element, which causes gene inactivation and recombination. However, the regulation and activation mechanisms of IS256 remain poorly understood. To describe how chronic lytic phage infection leads to extensive amplification of IS256 in E. faecalis and how antibiotic exposure is associated with amplification of IS256 in E. faecium during clinical human infection, Kirsch et al. [150] recently applied an IS256-specific deep sequencing approach. Comparative genomics assessment revealed that IS256 is predominantly expressed in hospital-acquired enterococcal isolates. IS256 mobility in E. faecalis is transcriptionally regulated by multiple mechanisms, indicating tight control of IS256 activation in the absence of selective pressure. The results show that rapid genome-scale transposition in enterococci is driven by stressors such as phages and antibiotic load. IS256 diversification may thereby illustrate how evolutionary selection mediates enterococcal genome evolution, ultimately leading to the development of dominant nosocomial lineages threatening human health.
Brown et al. have recently reported in an experimental mouse model setting that peritoneal inoculation of E. faecalis can result in sub-endothelial microlesions in the heart [116][151]. The study also showed a strong immune response to the infection, indicating that different inoculation routes may result in varying outcomes for both the host and the bacteria. E. faecalis invades the vascular endothelium to enter myocardial tissue and induce cell death [116]. Notably, E. faecalis lacks homologs of pneumococcal surface adhesin CbpA, pneumolysin (ply), and pyruvate oxidase (spxB), suggesting the involvement of other factors. However, it can produce reactive oxygen species (ROS) [152]. ROS release by E. faecalis may therefore also be involved in cell death and microlesion development. One protein that has been found to affect E. faecalis cardiac microlesion formation is a disulphide bond-forming (Dsb) protein called DsbA. Thioredoxins, such as DsbA, play a crucial role in various bacterial fitness and pathogenicity factors, including biofilm formation, cell division, virulence, motility, cell wall synthesis, and growth. Proteins with a highly expressed CXXC active site motif interact with the free thiol groups of substrate cysteines, catalysing a disulphide linkage. Gram-positive bacteria have a lesser understanding of oxidative protein folding than gram-negative bacteria [153].
References
- Hannachi, N.; Habib, G.; Camoin-Jau, L. Aspirin Effect on Staphylococcus aureus—Platelet Interactions During Infectious Endocarditis. Front. Med. 2019, 6, 217.
- Gaca, A.O.; Lemos, J.A. Adaptation to Adversity: The Intermingling of Stress Tolerance and Pathogenesis in Enterococci. Microbiol. Mol. Biol. Rev. 2019, 83, e00008-19.
- Fiore, E.; Van Tyne, D.; Gilmore, M.S. Pathogenicity of enterococci. Microbiol. Spectr. 2019, 7, 4.
- Goh, H.M.S.; Yong, M.H.A.; Chong, K.K.L.; Kline, K.A. Model systems for the study of Enterococcal colonization and infection. Virulence 2017, 8, 1525–1562.
- Ramsey, M.; Hartke, A.; Huycke, M. The Physiology and Metabolism of Enterococci. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014.
- Lebreton, F.; Willems, R.J.L.; Gilmore, M.S. Enterococcus Diversity, Origins in Nature, and Gut Colonization. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014.
- Boehm, A.B.; Sassoubre, L.M. Enterococci as Indicators of Environmental Fecal Contamination. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014.
- Nappi, F.; Singh, S.S.A.; Jitendra, V.; Fiore, A. Bridging Molecular and Clinical Sciences to Achieve the Best Treatment of Enterococcus faecalis Endocarditis. Microorganisms 2023, 11, 2604.
- Ch’ng, J.-H.; Chong, K.K.L.; Lam, L.N.; Wong, J.J.; Kline, K.A. Biofilm-associated infection by enterococci. Nat. Rev. Microbiol. 2018, 17, 82–94.
- Ramos, S.; Silva, V.; Dapkevicius, M.d.L.E.; Igrejas, G.; Poeta, P. Enterococci, from Harmless Bacteria to a Pathogen. Microorganisms 2020, 8, 1118.
- Holland, T.L.; Baddour, L.M.; Bayer, A.S.; Hoen, B.; Miro, J.M.; Fowler, V.G., Jr. Infective endocarditis. Nat. Rev. Dis. Primers 2016, 2, 16059.
- Nappi, F.; Martuscelli, G.; Bellomo, F.; Singh, S.S.A.; Moon, M.R. Infective Endocarditis in High-Income Countries. Metabolites 2022, 12, 682.
- Cahill, T.J.; Prendergast, B.D. Infective endocarditis. Lancet 2015, 387, 882–893.
- Barnes, A.M.T.; Dale, J.L.; Chen, Y.; Manias, D.A.; Quaintance, K.E.G.; Karau, M.K.; Kashyap, P.C.; Patel, R.; Wells, C.L.; Dunny, G.M. Enterococcus faecalis readily colonizes the entire gastrointestinal tract and forms biofilms in a germ-free mouse model. Virulence 2016, 8, 282–296.
- Mazzantini, D.; Calvigioni, M.; Celandroni, F.; Lupetti, A.; Ghelardi, E. Spotlight on the Compositional Quality of Probiotic Formulations Marketed Worldwide. Front. Microbiol. 2021, 12, 693973.
- Barnes, A.M.T.; Frank, K.L.; Dunny, G.M. Enterococcal Endocarditis: Hiding in Plain Sight. Front. Cell Infect. Microbiol. 2021, 11, 722482.
- Madsen, K.T.; Skov, M.N.; Gill, S.; Kemp, M. Virulence Factors Associated with Enterococcus faecalis Infective Endocarditis: A Mini Review. Open Microbiol. J. 2017, 11, 1–11.
- Kafil, H.S.; Mobarez, A.M. Spread of Enterococcal Surface Protein in Antibiotic Resistant Enterococcus faecium and Enterococcus faecalis isolates from Urinary Tract Infections. Open Microbiol. J. 2015, 9, 14–17.
- Frank, K.L.; Guiton, P.S.; Barnes, A.M.T.; Manias, D.A.; Chuang-Smith, O.N.; Kohler, P.L.; Spaulding, A.R.; Hultgren, S.J.; Schlievert, P.M.; Dunny, G.M. AhrC and Eep are biofilm infection-associated virulence factors in Enterococcus faecalis. Infect. Immun. 2013, 81, 1696–1708.
- Sillanpää, J.; Chang, C.; Singh, K.V.; Montealegre, M.C.; Nallapareddy, S.R.; Harvey, B.R.; Ton-That, H.; Murray, B.E. Contribution of individual Ebp Pilus subunits of Enterococcus faecalis OG1RF to pilus biogenesis, biofilm formation and urinary tract infection. PLoS ONE 2013, 8, e68813.
- Thurlow, L.R.; Thomas, V.C.; Narayanan, S.; Olson, S.; Fleming, S.D.; Hancock, L.E. Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect. Immun. 2010, 78, 4936–4943.
- Singh, K.V.; Nallapareddy, S.R.; Murray, B.E. Importance of the ebp (endocarditis- and biofilm-associated pilus) locus in the pathogenesis of Enterococcus faecalis ascending urinary tract infection. J. Infect. Dis. 2007, 195, 1671–1677.
- Rouchon, C.N.; Harris, J.; Zubair-Nizami, Z.; Weinstein, A.J.; Roky, M.; Frank, K.L. The Cationic Antimicrobial Peptide Activity of Lysozyme Reduces Viable Enterococcus faecalis Cells in Biofilms. Antimicrob. Agents Chemother. 2022, 66, e0233921.
- Qu, Q.; Chen, T.; He, P.; Geng, H.; Zeng, P.; Luan, G. Isolation and characterization of a novel lytic bacteriophage vB_Efm_LG62 infecting Enterococcus faecium. Virus Genes 2023, 59, 763–774.
- Holmberg, A.; Rasmussen, M. Mature biofilms of Enterococcus faecalis and Enterococcus faecium are highly resistant to antibiotics. Diagn. Microbiol. Infect. Dis. 2016, 84, 19–21.
- Nappi, F.; Schoell, T.; Spadaccio, C.; Acar, C.; da Costa, F.D.A. A Literature Review on the Use of Aortic Allografts in Modern Cardiac Surgery for the Treatment of Infective Endocarditis: Is There Clear Evidence or Is It Merely a Perception? Life 2023, 13, 1980.
- Andrewes, F.; Horder, T. A Study of the Streptococci Pathogenic for Man. Lancet 1906, 2, 708–713.
- Geraci, J.E.; Martin, W.J. Antibiotic therapy of bacterial endocarditis. VI. Subacute enterococcal endocarditis; clinical, pathologic and therapeutic consideration of 33 cases. Circulation 1954, 10, 173–194.
- Toh, C.C.S.; Bali, K.P. Natural History of Streptococcus faecalis Endocarditis. BMJ 1960, 2, 640–644.
- Jones, J.E.T. The experimental production of streptococcal endocarditis in the pig. J. Pathol. 1969, 99, 307–318.
- Durack, D.T.; Beeson, P.B.; Petersdorf, R.G. Experimental bacterial endocarditis. 3. Production and progress of the disease in rabbits. Br. J. Exp. Pathol. 1973, 54, 142–151.
- Schleifer, K.; Kilpper-Bälz, R.; Kraus, J.; Gehring, F. Relatedness and Classification of Streptococcus mutans and “Mutans-like” Streptococci. J. Dent. Res. 1984, 63, 1047–1050.
- Clewell, D.B. Movable genetic elements and antibiotic resistance in enterococci. Eur. J. Clin. Microbiol. Infect. Dis. 1990, 9, 90–102.
- E Murray, B. The life and times of the Enterococcus. Clin. Microbiol. Rev. 1990, 3, 46–65.
- Donati, L.; Scamazzo, F.; Gervasoni, M.; Magliano, A.; Stankov, B.; Fraschini, F. Infection and antibiotic therapy in 4000 burned patients treated in Milan, Italy, between 1976 and 1988. Burns 1993, 19, 345–348.
- Peng, M.Y.; Young, T.G.; Yang, C.H.; Chou, M.Y. Enterococcal bacteremia in a medical center. Zhonghua Yi Xue Za Zhi Chin. Med. J. 1994, 54, 306–311.
- Nicoletti, G.; Stefani, S. Enterococci: Susceptibility patterns and therapeutic options. Eur. J. Clin. Microbiol. Infect. Dis. 1995, 14 (Suppl. S1), S33–S37.
- de Vera, M.E.; Simmons, R.L. Antibiotic-resistant enterococci and the changing face of surgical infections. Arch. Surg. 1996, 131, 338–342.
- Gin, A.S.; Zhanel, G.G. Vancomycin-resistant enterococci. Ann. Pharmacother. 1996, 30, 615–624.
- Evers, S.; Quintiliani, R., Jr.; Courvalin, P. Genetics of glycopeptide resistance in enterococci. Microb. Drug Resist. 1996, 2, 219–223.
- Biavasco, F.; Miele, A.; Vignaroli, C.; Manso, E.; Lupidi, R.; Varaldo, P.E. Genotypic characterization of a nosocomial outbreak of VanA Enterococcus faecalis. Microb. Drug Resist. 1996, 2, 231–237.
- Shorrock, P.J.; A Lambert, P.; Aitchison, E.J.; Smith, E.G.; Farrell, I.D.; Gutschik, E. Serological response in Enterococcus faecalis endocarditis determined by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1990, 28, 195–200.
- Xu, Y.; Jiang, L.; E Murray, B.; Weinstock, G.M. Enterococcus faecalis antigens in human infections. Infect. Immun. 1997, 65, 4207–4215.
- Rich, R.L.; Kreikemeyer, B.; Owens, R.T.; LaBrenz, S.; Narayana, S.V.L.; Weinstock, G.M.; Murray, B.E.; Höök, M. Ace is a collagen-binding MSCRAMM from Enterococcus faecalis. J. Biol. Chem. 1999, 274, 26939–26945.
- Teng, F.; Jacques-Palaz, K.D.; Weinstock, G.M.; Murray, B.E. Evidence that the enterococcal polysaccharide antigen gene (epa) cluster is widespread in Enterococcus faecalis and influences resistance to phagocytic killing of E. faecalis. Infect. Immun. 2002, 70, 2010–2015.
- Ton-That, H.; Schneewind, O. Assembly of pili in Gram-positive bacteria. Trends Microbiol. 2004, 12, 228–234.
- Nallapareddy, S.R.; Singh, K.V.; Sillanpää, J.; Garsin, D.A.; Höök, M.; Erlandsen, S.L.; Murray, B.E. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Investig. 2006, 116, 2799–2807.
- Budzik, J.M.; Schneewind, O. Pili prove pertinent to enterococcal endocarditis. J. Clin. Investig. 2006, 116, 2582–2584.
- Kemp, K.D.; Singh, K.V.; Nallapareddy, S.R.; Murray, B.E. Relative contributions of Enterococcus faecalis OG1RF sortase-encoding genes, srtA and bps (srtC), to biofilm formation and a murine model of urinary tract infection. Infect. Immun. 2007, 75, 5399–5404.
- Scott, J.R.; Zähner, D. Pili with strong attachments: Gram-positive bacteria do it differently. Mol. Microbiol. 2006, 62, 320–330.
- Galli, D.; Wirth, R.; Wanner, G. Identification of aggregation substances of Enterococcus faecalis cells after induction by sex pheromones. An immunological and ultrastructural investigation. Arch. Microbiol. 1989, 151, 486–490.
- Olmsted, S.B.; Kao, S.M.; van Putte, L.J.; Gallo, J.C.; Dunny, G.M. Role of the pheromone-inducible surface protein Asc10 in mating aggregate formation and conjugal transfer of the Enterococcus faecalis plasmid pCF10. J. Bacteriol. 1991, 173, 7665–7672.
- Hirt, H.; Wanner, G.; Galli, D.; Wirth, R. Biochemical, immunological and ultrastructural characterization of aggregation substances encoded by Enterococcus faecalis sex-pheromone plasmids. Eur. J. Biochem. 1993, 211, 711–716.
- Dunny, G.M.; Leonard, B.A.; Hedberg, P.J. Pheromone-inducible conjugation in Enterococcus faecalis: Interbacterial and host-parasite chemical communication. J. Bacteriol. 1995, 177, 871–876.
- Leonard, B.A.; Bensing, B.A.; Hedberg, P.J.; Ruhfel, R.E.; Chung, J.W.; Dunny, G.M. Pheromone-inducible gene regulation and signalling for the control of aggregation substance expression in the conjugative plasmid pCF10. FEMS Microbiol. Lett. 1995, 85, 27–34.
- Nakayama, J.; Clewell, D.B.; Suzuki, A. Targeted disruption of the PD78 gene (traF) reduces pheromone-inducible conjugal transfer of the bacteriocin plasmid pPD1 in Enterococcus faecalis. FEMS Microbiol. Lett. 1995, 128, 283–288.
- Bae, T.; Kozlowicz, B.; Dunny, G.M. Two targets in pCF10 DNA for PrgX binding: Their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation. J. Mol. Biol. 2002, 315, 995–1007.
- Fernández-Hidalgo, N.; Escolà-Vergé, L.; Pericàs, J.M. Enterococcus faecalis endocarditis: What’s next? Futur. Microbiol. 2020, 15, 349–364.
- Llopis, J.; Muñoz, P.; Gálvez-Acebal, J.; Kestler, M.; Valerio, M.; Hernández-Meneses, M.; Cobo-Belaustegui, M.; Montejo, M.; Ojeda-Burgos, G.; Sousa-Regueiro, M.D.; et al. A Contemporary Picture of Enterococcal Endocarditis. J. Am. Coll. Cardiol. 2020, 75, 482–494.
- Escolà-Vergé, L.; Fernández-Hidalgo, N.; Larrosa, M.N.; Fernandez-Galera, R.; Almirante, B. Secular trends in the epidemiology and clinical characteristics of Enterococcus faecalis infective endocarditis at a referral center (2007–2018). Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 1137–1148.
- Bashore, T.M.; Turner, N.A. Addressing the Menace of Enterococcal Endocarditis. J. Am. Coll. Cardiol. 2020, 75, 495–497.
- Ramos-Martínez, A.; Domínguez, F.; Muñoz, P.; Marín, M.; Pedraz, Á.; Fariñas, M.C.; Tascón, V.; de Alarcón, A.; Rodríguez-García, R.; Miró, J.M.; et al. Clinical presentation, microbiology, and prognostic factors of prosthetic valve endocarditis. Lessons learned from a large prospective registry. PLoS ONE 2023, 18, e0290998.
- Herrera-Hidalgo, L.; Fernández-Rubio, B.; Luque-Márquez, R.; López-Cortés, L.E.; Gil-Navarro, M.V.; de Alarcón, A. Treatment of Enterococcus faecalis Infective Endocarditis: A Continuing Challenge. Antibiotics 2023, 12, 704.
- Parsek, M.R.; Fuqua, C. Biofilms 2003: Emerging themes and challenges in studies of surface-associated microbial life. J. Bacteriol. 2004, 186, 4502–4509.
- Häussler, S.; Parsek, M.R. Biofilms 2009: New perspectives at the heart of surface-associated microbial communities. J. Bacteriol. 2010, 192, 2941–2949.
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS 2013, 121, 1–58.
- Haussler, S.; Fuqua, C. Biofilms 2012: New discoveries and significant wrinkles in a dynamic field. J. Bacteriol. 2013, 195, 2947–2958.
- Visick, K.L.; Schembri, M.A.; Yildiz, F.; Ghigo, J.-M. Biofilms 2015: Multidisciplinary Approaches Shed Light into Microbial Life on Surfaces. J. Bacteriol. 2016, 198, 2553–2563.
- Høiby, N. A short history of microbial biofilms and biofilm infections. APMIS 2017, 125, 272–275.
- Fuqua, C.; Filloux, A.; Ghigo, J.-M.; Visick, K.L. Biofilms 2018: A diversity of microbes and mechanisms. J. Bacteriol. 2019, 201, e00118-19.
- Wang, T.; Zhang, R.; Chen, Z.; Cao, P.; Zhou, Q.; Wu, Q. A global bibliometric and visualized analysis of bacterial biofilm eradication from 2012 to 2022. Front. Microbiol. 2023, 14, 1287964.
- Săndulescu, O.; Săndulescu, M. Oral biofilms—Pivotal role in understanding microbes and their relevance to the human host. GERMS 2023, 13, 7–9.
- Hegstad, K.; Mikalsen, T.; Coque, T.M.; Werner, G.; Sundsfjord, A. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin. Microbiol. Infect. 2010, 16, 541–554.
- Paulsen, I.T.; Banerjei, L.; Myers, G.S.A.; Nelson, K.E.; Seshadri, R.; Read, T.D.; Fouts, D.E.; Eisen, J.A.; Gill, S.R.; Heidelberg, J.F.; et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 2003, 299, 2071–2074.
- Weigel, L.M.; Clewell, D.B.; Gill, S.R.; Clark, N.C.; McDougal, L.K.; Flannagan, S.E.; Kolonay, J.F.; Shetty, J.; Killgore, G.E.; Tenover, F.C. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571.
- Bourgogne, A.; Garsin, D.A.; Qin, X.; Singh, K.V.; Sillanpaa, J.; Yerrapragada, S.; Ding, Y.; Dugan-Rocha, S.; Buhay, C.; Shen, H.; et al. Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol. 2008, 9, R110.
- Palmer, K.L.; Carniol, K.; Manson, J.M.; Heiman, D.; Shea, T.; Young, S.; Zeng, Q.; Gevers, D.; Feldgarden, M.; Birren, B.; et al. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 2010, 192, 2469–2470.
- Kristich, C.J.; Chandler, J.R.; Dunny, G.M. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 2007, 57, 131–144.
- Kristich, C.J.; Manias, D.A.; Dunny, G.M. Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl. Environ. Microbiol. 2005, 71, 5837–5849.
- Kristich, C.J.; Nguyen, V.T.; Le, T.; Barnes, A.M.T.; Grindle, S.; Dunny, G.M. Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl. Environ. Microbiol. 2008, 74, 3377–3386.
- Ballering, K.S.; Kristich, C.J.; Grindle, S.M.; Oromendia, A.; Beattie, D.T.; Dunny, G.M. Functional genomics of Enterococcus faecalis: Multiple novel genetic determinants for biofilm formation in the core genome. J. Bacteriol. 2009, 191, 2806–2814.
- Frank, K.L.; Barnes, A.M.T.; Grindle, S.M.; Manias, D.A.; Schlievert, P.M.; Dunny, G.M. Use of recombinase-based in vivo expression technology to characterize Enterococcus faecalis gene expression during infection identifies in vivo-expressed antisense RNAs and implicates the protease Eep in pathogenesis. Infect. Immun. 2012, 80, 539–549.
- Frank, K.L.; Vergidis, P.; Brinkman, C.L.; Quaintance, K.E.G.; Barnes, A.M.T.; Mandrekar, J.N.; Schlievert, P.M.; Dunny, G.M.; Patel, R. Evaluation of the Enterococcus faecalis Biofilm-Associated Virulence Factors AhrC and Eep in Rat Foreign Body Osteomyelitis and In Vitro Biofilm-Associated Antimicrobial Resistance. PLoS ONE 2015, 10, e0130187.
- Leuck, A.-M.; Johnson, J.R.; Dunny, G.M. A widely used in vitro biofilm assay has questionable clinical significance for enterococcal endocarditis. PLoS ONE 2014, 9, e107282.
- Colomer-Winter, C.; Gaca, A.O.; Chuang-Smith, O.N.; Lemos, J.A.; Frank, K.L. Basal levels of (p)ppGpp differentially affect the pathogenesis of infective endocarditis in Enterococcus faecalis. Microbiology 2018, 164, 1254–1265.
- Dunny, G.M.; Hancock, L.E.; Shankar, N. Enterococcal Biofilm Structure and Role in Colonization and Disease. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. Available online: https://www.ncbi.nlm.nih.gov/books/NBK190433 (accessed on 3 February 2024).
- Manias, D.A.; Dunny, G.M. Expression of Adhesive Pili and the Collagen-Binding Adhesin Ace Is Activated by ArgR Family Transcription Factors in Enterococcus faecalis. J. Bacteriol. 2018, 200, e00269-18.
- Nappi, F.; Spadaccio, C.; Dreyfus, J.; Attias, D.; Acar, C.; Bando, K. Mitral endocarditis: A new management framework. J. Thorac. Cardiovasc Surg. 2018, 156, 1486–1495.e4.
- Arthur, M.; Reynolds, P.E.; Depardieu, F.; Evers, S.; Dutka-Malen, S.; Quintiliani, R., Jr.; Courvalin, P. Mechanisms of glycopep- tide resistance in enterococci. J. Infect. 1996, 32, 11–16.
- Arthur, M.; Depardieu, F.; Gerbaud, G.; Galimand, M.; Leclercq, R.; Courvalin, P. The VanS sensor negatively controls VanR-mediated transcriptional activation of glycopeptide resistance genes of Tn 1546 and related elements in the absence of induction. J. Bacteriol. 1997, 179, 97–106.
- Bugg, T.D.H.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415.
- Mohamed, J.A.; Huang, D.B. Biofilm formation by enterococci. J. Med. Microbiol. 2007, 56 Pt 12, 1581–1588.
- Mohamed, J.A.; Teng, F.; Nallapareddy, S.R.; Murray, B.E. Pleiotrophic effects of 2 Enterococcus faecalis sagA–like genes, salA and salB, which encode proteins that are antigenic during human infection, on biofilm formation and binding to collagen type i and fibronectin. J. Infect. Dis. 2006, 193, 231–240.
- Milbrandt, E. A novel source of enterococcal endocarditis. Clin. Cardiol. 1998, 21, 123–126.
- Manoil, D.; Cerit, E.E.; Fang, H.; Durual, S.; Brundin, M.; Belibasakis, G.N. Profiling Antibiotic Susceptibility among Distinct Enterococcus faecalis Isolates from Dental Root Canals. Antibiotics 2023, 13, 18.
- Pandova, M.; Kizheva, Y.; Tsenova, M.; Rusinova, M.; Borisova, T.; Hristova, P. Pathogenic Potential and Antibiotic Susceptibility: A Comprehensive Study of Enterococci from Different Ecological Settings. Pathogens 2023, 13, 36.
- Mancuso, G.; Midiri, A.; Gerace, E.; Marra, M.; Zummo, S.; Biondo, C. Urinary Tract Infections: The Current Scenario and Future Prospects. Pathogens 2023, 12, 623.
- Arias, C.A.; Murray, B.E. The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 2012, 10, 266–278.
- Khan, Z.; Siddiqui, N.; Saif, M.W. Enterococcus Faecalis Infective Endocarditis and Colorectal Carcinoma: Case of New Association Gaining Ground. Gastroenterol. Res. 2018, 11, 238–240.
- Jahansepas, A.; Aghazadeh, M.; Rezaee, M.A.; Hasani, A.; Sharifi, Y.; Aghazadeh, T.; Mardaneh, J. Occurrence of Enterococcus faecalis and Enterococcus faecium in Various Clinical Infections: Detection of Their Drug Resistance and Virulence Determinants. Microb. Drug Resist. 2018, 24, 76–82.
- Coccitto, S.N.; Cinthi, M.; Simoni, S.; Pocognoli, A.; Zeni, G.; Mazzariol, A.; Morroni, G.; Mingoia, M.; Giovanetti, E.; Brenciani, A.; et al. Genetic analysis of vancomycin-variable Enterococcus faecium clinical isolates in Italy. Eur. J. Clin. Microbiol. Infect. Dis. 2024.
- Dubin, K.; Pamer, E.G. Enterococci and Their Interactions with the Intestinal Microbiome. Microbiol. Spectr. 2017, 5, 5–6.
- Hendrickx, A.P.A.; Top, J.; Bayjanov, J.R.; Kemperman, H.; Rogers, M.R.C.; Paganelli, F.L.; Bonten, M.J.M.; Willems, R.J.L. Antibiotic-Driven Dysbiosis Mediates Intraluminal Agglutination and Alternative Segregation of Enterococcus faecium from the Intestinal Epithelium. mBio 2015, 6, e01346-15.
- Wells, C.L.; Jechorek, R.P.; Erlandsen, S.L. Evidence for the translocation of Enterococcus faecalis across the mouse intestinal tract. J. Infect. Dis. 1990, 162, 82–90.
- Qin, X.; Singh, K.V.; Weinstock, G.M.; Murray, B.E.; Klee, S.R.; Nassif, X.; Kusecek, B.; Merker, P.; Beretti, J.-L.; Achtman, M.; et al. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect. Immun. 2000, 68, 2082–2095.
- Zeng, J.; Teng, F.; Weinstock, G.M.; Murray, B.E. Translocation of Enterococcus faecalis Strains across a monolayer of polarized human enterocyte-like T84 cells. J. Clin. Microbiol. 2004, 42, 1149–1154.
- Zeng, J.; Teng, F.; Murray, B.E. Gelatinase Is Important for translocation of Enterococcus faecalis across polarized human enterocyte-like T84 cells. Infect. Immun. 2005, 73, 1606–1612.
- Archambaud, C.; Derré-Bobillot, A.; Lapaque, N.; Rigottier-Gois, L.; Serror, P. Intestinal translocation of enterococci requires a threshold level of enterococcal overgrowth in the lumen. Sci. Rep. 2019, 9, 8926.
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161.
- Fine, R.L.; Vieira, S.M.; Gilmore, M.S.; Kriegel, M.A. Mechanisms and consequences of gut commensal translocation in chronic diseases. Gut Microbes 2020, 11, 217–230.
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780.
- Tie, Y.; Huang, Y.; Chen, R.; Li, L.; Chen, M.; Zhang, S. Current insights on the roles of gut microbiota in inflammatory bowel disease-associated extra-intestinal manifestations: Pathophysiology and therapeutic targets. Gut Microbes 2023, 15, 2265028.
- Knoop, K.A.; McDonald, K.G.; Kulkarni, D.H.; Newberry, R.D. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut 2016, 65, 1100–1109.
- Kulkarni, D.H.; Rusconi, B.; Floyd, A.N.; Joyce, E.L.; Talati, K.B.; Kousik, H.; Alleyne, D.; Harris, D.L.; Garnica, L.; McDonough, R.; et al. Gut microbiota induces weight gain and inflammation in the gut and adipose tissue independent of manipulations in diet, genetics, and immune development. Gut Microbes 2023, 15, 2284240.
- Hill, C.A.; Casterline, B.W.; Valguarnera, E.; Hecht, A.L.; Shepherd, E.S.; Sonnenburg, J.L.; Wardenburg, J.B. Bacteroides fragilis toxin expression enables lamina propria niche acquisition in the developing mouse gut. Nat. Microbiol. 2024, 9, 85–94.
- Brown, A.; Singh, K.V.; Cruz, M.R.; Kaval, K.G.; Francisco, L.E.; Murray, B.E.; A Garsin, D. Cardiac Microlesions Form During Severe Bacteremic Enterococcus faecalis Infection. J. Infect. Dis. 2021, 223, 508–516.
- Nappi, F.; Spadaccio, C.; Acar, C. Use of allogeneic tissue to treat infective valvular disease: Has everything been said? J. Thorac. Cardiovasc. Surg. 2017, 153, 824–828.
- Cabiltes, I.; Coghill, S.; Bowe, S.J.; Athan, E. Enterococcal bacteraemia ‘silent but deadly’: A population-based cohort study. Intern. Med. J. 2019, 50, 434–440.
- Ambrosioni, J.; Muñoz, P.; de Alarcón, A.; Kestler, M.; Mari-Hualde, A.; Moreno, A.; Rodríguez-Álvarez, R.; Ojeda-Burgos, G.; Gálvez-Acebal, J.; Hidalgo-Tenorio, C.; et al. Prevalence of Colorectal Neoplasms Among Patients With Enterococcus faecalis Endocarditis in the GAMES Cohort (2008–2017). Mayo Clin. Proc. 2021, 96, 132–146.
- Pasquereau-Kotula, E.; Martins, M.; Aymeric, L.; Dramsi, S. Significance of Streptococcus gallolyticus subsp. gallolyticus Association With Colorectal Cancer. Front. Microbiol. 2018, 9, 614.
- Jans, C.; Boleij, A. The Road to Infection: Host-Microbe Interactions Defining the Pathogenicity of Streptococcus bovis/Streptococcus equinus Complex Members. Front. Microbiol. 2018, 9, 603.
- Aymeric, L.; Donnadieu, F.; Mulet, C.; du Merle, L.; Nigro, G.; Saffarian, A.; Bérard, M.; Poyart, C.; Robine, S.; Regnault, B.; et al. Colorectal cancer specific conditions promote Streptococcus gallolyticus gut colonization. Proc. Natl. Acad. Sci. USA 2017, 115, E283–E291.
- Taylor, J.C.; Gao, X.; Xu, J.; Holder, M.; Petrosino, J.; Kumar, R.; Liu, W.; Höök, M.; Mackenzie, C.; Hillhouse, A.; et al. A type VII secretion system of Streptococcus gallolyticus subsp. gallolyticus contributes to gut colonization and the development of colon tumors. PLoS Pathog. 2021, 17, e1009182.
- Taylor, J.C.; Kumar, R.; Xu, J.; Xu, Y. A pathogenicity locus of Streptococcus gallolyticus subspecies gallolyticus. Sci. Rep. 2023, 13, 6291.
- Stanley, D.; Mason, L.J.; E Mackin, K.; Srikhanta, Y.N.; Lyras, D.; Prakash, M.D.; Nurgali, K.; Venegas, A.; Hill, M.D.; Moore, R.J.; et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 2016, 22, 1277–1284.
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019.
- Claes, J.; Ditkowski, B.; Liesenborghs, L.; Veloso, T.R.; Entenza, J.M.; Moreillon, P.; Vanassche, T.; Verhamme, P.; Hoylaerts, M.F.; Heying, R. Assessment of the Dual Role of Clumping Factor A in S. Aureus Adhesion to Endothelium in Absence and Presence of Plasma. Arthritis Res. Ther. 2018, 118, 1230–1241.
- Ko, Y.-P.; Kang, M.; Ganesh, V.K.; Ravirajan, D.; Li, B.; Höök, M. Coagulase and Efb of Staphylococcus aureus Have a Common Fibrinogen Binding Motif. mBio 2016, 7, e01885-15.
- Pappelbaum, K.I.; Gorzelanny, C.; Grässle, S.; Suckau, J.; Laschke, M.W.; Bischoff, M.; Bauer, C.; Schorpp-Kistner, M.; Weidenmaier, C.; Schneppenheim, R.; et al. Ultralarge von willebrand factor fibers mediate luminal Staphylococcus aureus adhesion to an intact endothelial cell layer under shear stress. Circulation 2013, 128, 50–59.
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241.
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218.
- Nappi, F.; Bellomo, F.; Singh, S.S.A. Insights into the Role of Neutrophils and Neutrophil Extracellular Traps in Causing Cardiovascular Complications in Patients with COVID-19: A Systematic Review. J. Clin. Med. 2022, 11, 2460.
- Nappi, F.; Iervolino, A.; Singh, S.S.A. Thromboembolic Complications of SARS-CoV-2 and Metabolic Derangements: Suggestions from Clinical Practice Evidence to Causative Agents. Metabolites 2021, 11, 341.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Nappi, F.; Bellomo, F.; Singh, S.S.A. Worsening Thrombotic Complication of Atherosclerotic Plaques Due to Neutrophils Extracellular Traps: A Systematic Review. Biomedicines 2023, 11, 113.
- Nappi, F.; Nappi, P.; Gambardella, I.; Singh, S.S.A. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites 2022, 12, 889.
- Morrell, C.N.; Hilt, Z.T.; Pariser, D.N.; Maurya, P. PAD4 and von Willebrand Factor Link Inflammation and Thrombosis. Circ. Res. 2019, 125, 520–522.
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ. Res. 2019, 125, 507–519.
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. J. Clin. Med. 2019, 8, 2072.
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-dependent coagulation activation in sepsis. Front. Immunol. 2021, 12, 641750.
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737.
- Braï, M.A.; Hannachi, N.; El Gueddari, N.; Baudoin, J.-P.; Dahmani, A.; Lepidi, H.; Habib, G.; Camoin-Jau, L. The Role of Platelets in Infective Endocarditis. Int. J. Mol. Sci. 2023, 24, 7540.
- Misfeldt, A.M.; Boyle, S.C.; Tompkins, K.L.; Bautch, V.L.; Labosky, P.A.; Baldwin, H.S. Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev. Biol. 2009, 333, 78–89.
- Dyer, L.; Patterson, C. Development of the endothelium: An emphasis on heterogeneity. Semin. Thromb. Hemost. 2010, 36, 227–235.
- Harris, I.S.; Black, B.L. Development of the Endocardium. Pediatr. Cardiol. 2010, 31, 391–399.
- Milgrom-Hoffman, M.; Harrelson, Z.; Ferrara, N.; Zelzer, E.; Evans, S.M.; Tzahor, E. The heart endocardium is derived from vascular endothelial progenitors. Development 2011, 138, 4777–4787.
- Borasch, K.; Richardson, K.; Plendl, J. Cardiogenesis with a focus on vasculogenesis and angiogenesis. Anat. Histol. Embryol. 2020, 49, 643–655.
- Perez, M.; Calles-Enríquez, M.; del Rio, B.; Ladero, V.; Martín, M.C.; Fernández, M.; Alvarez, M.A. IS256 abolishes gelatinase activity and biofilm formation in a mutant of the nosocomial pathogen Enterococcus faecalis V583. Can. J. Microbiol. 2015, 61, 517–519.
- Ali, L.; Goraya, M.U.; Arafat, Y.; Ajmal, M.; Chen, J.-L.; Yu, D. Molecular Mechanism of Quorum-Sensing in Enterococcus faecalis: Its Role in Virulence and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 960.
- Kirsch, J.M.; Ely, S.; Stellfox, M.E.; Hullahalli, K.; Luong, P.; Palmer, K.L.; Van Tyne, D.; Duerkop, B.A. Targeted IS-element sequencing uncovers transposition dynamics during selective pressure in enterococci. PLoS Pathog. 2023, 19, e1011424.
- Brown, A.O.; Garsin, D.A. The pathogenesis of cardiac microlesion formation during severe bacteremic infection. PLoS Pathog. 2020, 16, e1009021.
- Wang, X.; Huycke, M.M. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 2007, 132, 551–561.
- Reardon-Robinson, M.E.; Ton-That, H. Disulfide-Bond-Forming Pathways in Gram-Positive Bacteria. J. Bacteriol. 2016, 198, 746–754.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
621
Revisions:
2 times
(View History)
Update Date:
26 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No