Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marisa Rego Encarnação | -- | 2483 | 2024-03-22 18:33:03 | | | |
| 2 | Peter Tang | + 5 word(s) | 2488 | 2024-03-25 02:28:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Encarnação, M.; Ribeiro, I.; David, H.; Coutinho, M.F.; Quelhas, D.; Alves, S. Niemann–Pick Type C—Leaky Variants and Alternative Transcripts. Encyclopedia. Available online: https://encyclopedia.pub/entry/56462 (accessed on 27 July 2026).
Encarnação M, Ribeiro I, David H, Coutinho MF, Quelhas D, Alves S. Niemann–Pick Type C—Leaky Variants and Alternative Transcripts. Encyclopedia. Available at: https://encyclopedia.pub/entry/56462. Accessed July 27, 2026.
Encarnação, Marisa, Isaura Ribeiro, Hugo David, Maria Francisca Coutinho, Dulce Quelhas, Sandra Alves. "Niemann–Pick Type C—Leaky Variants and Alternative Transcripts" Encyclopedia, https://encyclopedia.pub/entry/56462 (accessed July 27, 2026).
Encarnação, M., Ribeiro, I., David, H., Coutinho, M.F., Quelhas, D., & Alves, S. (2024, March 22). Niemann–Pick Type C—Leaky Variants and Alternative Transcripts. In Encyclopedia. https://encyclopedia.pub/entry/56462
Encarnação, Marisa, et al. "Niemann–Pick Type C—Leaky Variants and Alternative Transcripts." Encyclopedia. Web. 22 March, 2024.
Copy Citation
Niemann–Pick type C (NPC, ORPHA: 646) is a neuro-visceral, psychiatric disease caused predominantly by pathogenic variants in the NPC1 gene or seldom in NPC2. The rarity of the disease, and its wide range of clinical phenotypes and ages of onset, turn the diagnosis into a significant challenge. Other than the detailed clinical history, the typical diagnostic work-up for NPC includes the quantification of pathognomonic metabolites.
Niemann–Pick type C
splicing variants
leaky variants
NPC1 gene
molecular diagnosis
1. Introduction
Lysosomal storage disorders (LSDs) are a group of about 70 inherited diseases, most of which are quite rare and present with vast clinical heterogeneity, ranging from severe, early-onset diseases to milder forms, of later onset. This remarkable variability may be observed not only between different diseases from the same group but also—and most importantly—amongst patients suffering from the same exact disease. Overall, this clinical heterogeneity has a direct impact on their diagnosis. Over the past several years, the number of available treatments for patients with LSDs has rapidly increased, namely, enzyme replacement and substrate reduction therapies, the use of molecular chaperones, gene therapy, and bone marrow transplant, among others [1]. Nevertheless, molecular diagnosis is the ultimate and essential step to provide access to therapy. The identification of biallelic nonsense and frameshift variants, as well as missense variants in conserved regions, provides a straightforward direct gene target analysis. Nevertheless, next-generation sequencing (NGS)-targeted panels for LSD-associated genes or other NGS methodologies provide a quick way to identify the molecular defect underlying diseases with such clinical variability [2]. However, in specific cases, the molecular diagnosis timeline can be even longer, when such pathogenic variants affect splicing and mRNA processing. These situations represent an additional challenge, with the identification and effect prediction of abnormal transcripts. In addition, some naturally spliced forms can raise another layer of difficulty and even mimic the molecular defect and its impact on splicing. The IDS, GNPTAB, and NPC1 genes—whose pathogenic variants underly Mucopolysaccharidosis type II, Mucolipidosis types II or III, and Niemann–Pick type C, respectively—are some examples of LSD-associated genes with naturally occurring spliced forms already reported in the databases (https://www.uniprot.org/uniprotkb/P22304/entry#sequences (accessed on 28 September 2023)); (https://www.uniprot.org/uniprotkb/Q3T906/entry#sequences (accessed on 28 September 2023)); and (https://www.uniprot.org/uniprotkb/O15118/entry#sequences (accessed on 28 September 2023)).
Niemann–Pick type C (NPC, ORPHA: 646) in particular is a devastating neurodegenerative LSD, caused by loss-of-function variants in either the NPC1 gene (in approximately 95% of cases) [3] or the NPC2 gene (in 5% of cases). Analysis of next-generation sequencing (NGS) data sets indicates that the incidence rate of NPC for the classical clinical manifestations is ~1:90,000 but suggests that, for the late-onset phenotype or variant forms, the frequency might be higher [4].
Overall, the wide range of clinical phenotypes and the different ages of onset it may present with, together with the rarity of the disease and the fact it may be caused by mutations in two different genes, make its diagnosis a significant challenge. At a clinical level, NPC’s infantile forms present varying degrees of neurologic involvement and frequently present visceral manifestations, such as splenomegaly, hepatomegaly, neonatal jaundice, and hyperbilirubinemia [5][6]. Adolescent- or adult-onset NPC, on the other hand, presents with varying combinations of progressive neurologic deficits, e.g., ataxia, dystonia and/or dementia, vertical supranuclear gaze palsy (VSGP), or major psychiatric illness, including schizophrenia, depression, and psychosis, among others [6].
That is why a definitive NPC diagnosis must rely on additional laboratorial analyses. The classical method of establishing a NPC diagnosis relies on the filipin staining of cultured fibroblasts from skin biopsies [7]. This is a microscopy-based test that takes advantage of the fact that filipin specifically binds to unesterified cholesterol, allowing the evaluation of cholesterol accumulation in the perinuclear vesicular compartments [8]. This rationale is consistent with the current assumption that the impaired egress of cholesterol from the late endosome/lysosome (LE/L) is a key element of NPC pathogenesis. Nevertheless, even the most severely affected patients may fail to be diagnosed through this method [9][10]. In fact, patients with proven NPC disease may present with variable filipin patterns, from typical “classical” or “intermediate” to “atypical” or “variant” ones, which fail to be classified as a NPC by filipin staining alone. Recent advances in the field are actively contributing to an increase in the detection of NPC patients. Among those advances is the development of rapid and reliable biomarkers, including oxysterols [11][12][13], lysosphingomyelin derivatives [14][15], and bile acids [16][17], even though none of them are specific to NPC [18]. However, N-palmitoyl-O-phosphocholineserine, (PPCS, previously known as lysosphingomyelin-509) has been shown to be elevated in the plasma and dried blood spots of NPC patients [19][20]. But, these novel biomarkers are not the sole contributors to the increased recognition of this disorder and its more expedited diagnosis. The increased availability of NGS has also contributed to the update of the overall NPC diagnostic algorithm while actively contributing to an increase in the number of positive molecular NPC diagnoses. Currently, there are a number of fully described diagnostic workflows for NPC [18], which may slightly vary between different labs depending on the tests each of them has available. However, NPC1 and NPC2 molecular analysis is mandatory in all of them and usually represents the ultimate step towards diagnosis [21]. Indeed, a rapid molecular diagnosis of a potential NPC patient is essential, not just for swift access to available therapies (currently miglustat is the only one approved within the European Union) but also to slow the progression of the disease and ultimately because it is the sole method of offering prenatal diagnosis to affected families [22].
2. A Brief Overview of the Diagnostic Workflow for Niemann-Pick Type C
2.1. The Picture in Black and White: Standard Workflows and Straightforward Diagnoses
In general, following a suspicious timeline of clinical manifestation and/or a biomarker profile consistent with NPC, the next step is the NPC1 and NPC2 sequencing of the index case (Figure 1) and subsequent segregation studies of the parents [18]. The NPC1 gene (MIM# 607623) comprises 25 exons and over 600 disease-causing variants have been reported to date [23], most of which encode missense alleles. For the NPC2 gene (MIM# 601015), thirty-four disease-causing variants have been described and four of them are splicing (https://my.qiagendigitalinsights.com/, HGMD Professional 2023.3, accessed on 17 October 2023). Among the most common NPC1 pathogenic variants are p.Ile1061Thr, found in 20% of patients of Western European descent [24], and p.Pro1007Ala, associated with milder forms of the disease [25][26]. In Portugal, however, the most frequent disease-causing variant is the missense p.Ala1035Val, which accounts for 15–20% of the affected cases (unpublished data transmitted by Quelhas D and Ribeiro I); it was recently reported as the most common in patients from Latin America [27].
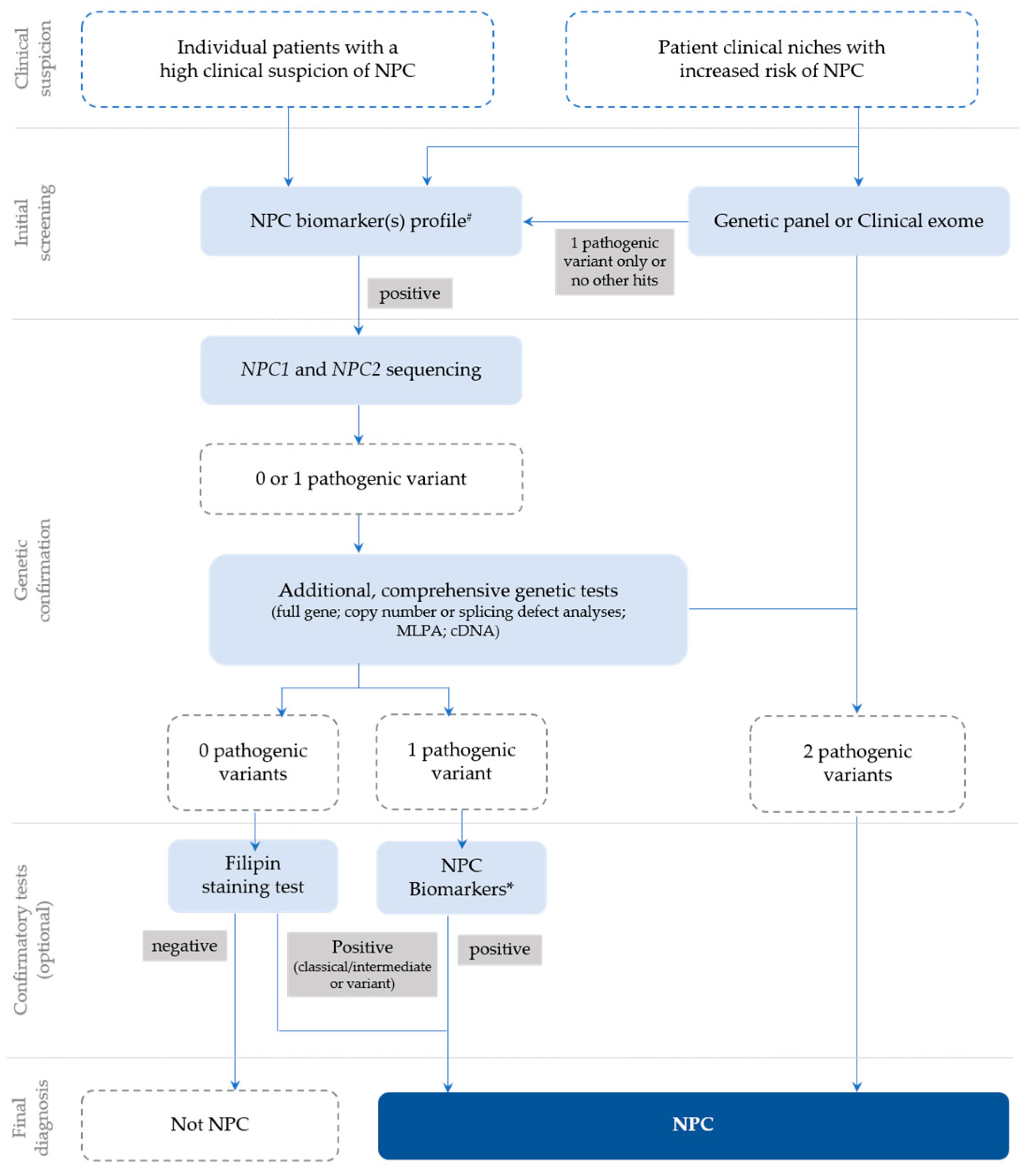
Figure 1. Recommendations for the detection and diagnosis of NPC, based on Patterson et al. [18] with slight updates to accommodate the most recent technologies, which are now commonly used for diagnostic purposes (e.g., clinical exome), as well as the current nomenclature. # Negative biomarkers may be suggestive that the diagnosis is not NPC; * Biomarker(s) profiling (if not initially conducted) or extended biomarker(s) profiling (in addition to those already conducted).
However, the highly polymorphic nature of NPC1 can muddle diagnostic conclusions and turn the interpretation of novel variants of unknown significance (VUSs) into a challenge. In addition, cDNA sequencing is necessary to address mRNA processing in the presence of silent variants, or other VUSs, including missense variants near the (exonic or intronic) splicing regions.
More specifically, the cDNA analysis of exonic variants may help confirm the pathogenic effect of variants predicted to affect splice sites [28]. Several splice-site pathogenic variants have been identified in NPC and in many other LSDs [29]. In some instances, these variants do not allow the generation of functional mRNAs [30]. However, they are leaky and frequently produce a small percentage of correctly spliced and translated transcripts, leading to attenuated phenotypic expression of the disease [31].
Whenever conventional gDNA analysis leads to a single variant identification, the genetic study focuses on detecting the second damaging variant. For this reason, complementary studies, such as multiplex ligation-dependent probe amplification (MLPA) in gDNA to cover intragenic deletions or duplications or cDNA sequencing, may also be required for proper diagnosis of NPC [18][22]. As straightforward as this approach may sound, reaching a conclusive molecular diagnosis of NPC may, in some cases, be harder than it seems.
2.2. The Grayscale Image
Among the confounding factors that can either hinder or delay a definitive diagnosis of NPC is the presence of genetic variants affecting the normal NPC1 and NPC2 splicing patterns.
Several pathogenic variants affecting both NPC1 and NPC2 mRNA splicing, occurring in intronic and exonic regions, have already been described [32]. Although quite rare, three pathogenic intronic variants have been described in the NPC2 gene [12][28][33]. One additional variant affecting splicing was found in both healthy controls and patients [4].
Interestingly, missense variants, such as the c.1553G>A (p.Arg518Gln), were proven to have an additional impact on the splicing mechanism in the NPC1 gene, as long as they occur in the coding exons’ splicing regulatory sequences [28][34][35][36].
Following a combined approach (gDNA and cDNA studies), the researchers have previously proven the impact of a silent variant in the NPC1 gene that leads to exon skipping—p.Val562= (Figure 2) [37]. This variant is located in Exon 11 and was initially reported in Spanish NPC patients and classified, at that time, as a VUS or polymorphism, after a genomic DNA study [38].

Figure 2. Schematic representation of the silent variants in the NPC1 exonic region affecting splicing and the effect on splicing based on in silico predictions (Human Splicing Finder—HSF and EX-SKIP tools and Maxent). P.Val562= localization on Exon 11 (red) and the effect on splicing based on in silico predictions. EX-SKIP compares the Exonic Splicing Enhancer (ESE)/Exonic Splicing Silencer (ESS) profile of a wild type (WT) and a mutated allele to determine if a specific exonic variant increases the chance of exon skipping. It calculates the total number of ESSs, ESEs, and their ratio. The p.Val562= mutant is associated with a change in the ESE/ESS ratio, which is compatible with a higher chance of exon skipping than in the WT allele. In addition, the HSF (a tool to predict the effects of pathogenic variants on splicing signals or to identify splicing motifs in any human sequence) predicts that the p.Val562= mutant leads to the creation of an ESS site. It involves the cDNA sequences CTTGTAAT (orange) [39] and CTTGTA (yellow) [40], which might be associated with a potential alteration of splicing. In the case of the silent variant p.Val562=, functional cDNA analysis was performed [19], confirming the bioinformatic prediction of Exon 11 skippings.
In the previous study, cDNA analysis of the affected patient and his mother (heterozygous carrier) made it possible to identify a transcript with the skipping of Exon 11. This caused a shift in the reading frame and the emergence of a premature termination codon [37], leading to its reclassification as a disease-causing variant. Importantly, the p.Val562= variant was found not only in three independent Portuguese families but also in previously reported Spanish [38] and French patients [41]. In a French cohort, this variant was reported in heterozygosity with the p.Ile1061Thr in two siblings [41][42]; however, its functional consequence was not ascertained. Despite the sequencing of five overlapping NPC1 cDNA fragments in the two siblings carrying the p.Val562=, the pathogenic effect of the variant was considered unknown [41]. The most likely explanation is the degradation of the aberrant transcript by NMD. There is no information about the frequency of this variant in gnomAD. Looking to other repositories, this variant is only reported in a database from Tubingen University (NPC-db2; https://medgen.medizin.uni-tuebingen.de/NPC-db2/search.php (accessed on 28 September 2023))—it was found in one patient but not in the controls. No information was provided regarding the homo- or heterozygosity of that patient; however, both in the literature and in the researchers' cohort, only heterozygous patients were identified.
Another example of a NPC-causing variant associated with complex mRNA processing is the c.190+5G>A variant. This particular variant is located not in NPC1, the most obvious candidate to harbor a disease-causing mutation, but in Intron 2 of the NPC2 gene. Again, this variant seems to be associated with a milder clinical course since both reported patients—two siblings homozygous for this variant—presented with a juvenile onset of the neurological disease and prolonged survival. A more detailed study showed that this splice variant generated multiple abnormal mRNAs [43]. However, in fibroblasts, a very small proportion of the correctly spliced transcript was also observed. Although this was not sufficient in producing enough NPC2 protein for Western Blot detection, the presence of low levels of functional protein presumably accounts for the milder clinical course. The question of whether different tissues could display variable levels of abnormally/normally spliced RNA transcribed from the c.190+5G>A variant can also be raised.
Ideally, however, these variants should be easily detected whenever an adequate cDNA analysis of any of the involved genes is performed. Still, that is not always the case, as the researchers will demonstrate with a few practical examples.
3. Variants in the NPC1 Gene That Affect Splicing
Pathogenic variants that affect pre-mRNA splicing account for at least 15% of disease-causing mutations [44]. Most of these variants affect 5′ and 3′ss, the polypyrimidine tract, the branch-point sequence, and also cis-acting elements (exonic/intronic splicing enhancers and silencers). Other variants create novel splicing sequences deeply within introns, causing the abnormal inclusion of intron sequences. All of these variants lead to the production of abnormal transcripts that usually contain PTCs and are degraded by nonsense-mediated mRNA decay (NMD) [45]. Even exonic variants (missense and synonymous) may affect splicing, having a completely different effect from what was expected [46]. Therefore, both gDNA and cDNA should be analyzed.
The NPC1 gene contains 25 exons and spans 55 kb from the base pairs (23, 531, 442) to 23, 586, 506 on the reverse strand of chromosome 18 at 18q11.2 (NC_000018.10); here the researchers report on 53 published pathogenic variants affecting splicing.
Seven of them are exonic, two are synonymous, five are missense, and forty-six are intronic variants, mainly affecting the 3’ss and the 5’ss; however, there are also variants reported in the branch point as well as deep-intronic variants.
Altogether, these observations call attention to the need for extensive mRNA studies in NPC1, or even NPC2, to establish a definitive NPC diagnosis. In this context, the presence of an alternatively spliced transcript may be somewhat confusing and even mask or mimic a real pathogenic variant that impacts splicing only in NPC patients and NPC1 variant carriers.
As for tissue-specific differences in the relative abundance of the two NPC1 splice isoforms, the researchers observed a higher expression of the spliced isoform in fibroblasts than in blood. In fact, in the genotype-tissue expression project (GTEx), which studies tissue-specific gene expression and regulation in fibroblasts, they quantified 30 fragments per kilobase of exon per million fragments mapped; meanwhile, in whole blood, only 17 fragments were quantified (https://www.genecards.org/cgi-bin/carddisp.pl?gene=NPC1&keywords=npc1#expression (accessed on 28 September 2023)).
References
- Beck, M. Treatment Strategies for Lysosomal Storage Disorders. Dev. Med. Child Neurol. 2018, 60, 13–18.
- Encarnação, M.; Coutinho, M.F.; Silva, L.; Ribeiro, D.; Ouesleti, S.; Campos, T.; Santos, H.; Martins, E.; Cardoso, M.T.; Vilarinho, L.; et al. Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants. Int. J. Mol. Sci. 2020, 21, 6355.
- Millat, G.; Baïlo, N.; Molinero, S.; Rodriguez, C.; Chikh, K.; Vanier, M.T. Niemann-Pick C Disease: Use of Denaturing High Performance Liquid Chromatography for the Detection of NPC1 and NPC2 Genetic Variations and Impact on Management of Patients and Families. Mol. Genet. Metab. 2005, 86, 220–232.
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High Incidence of Unrecognized Visceral/Neurological Late-Onset Niemann-Pick Disease, Type C1, Predicted by Analysis of Massively Parallel Sequencing Data Sets. Genet. Med. 2016, 18, 41–48.
- Pineda, M.; Mengel, E.; Jahnová, H.; Héron, B.; Imrie, J.; Lourenço, C.M.; van der Linden, V.; Karimzadeh, P.; Valayannopoulos, V.; Jesina, P.; et al. A Suspicion Index to Aid Screening of Early-Onset Niemann-Pick Disease Type C (NP-C). BMC Pediatr. 2016, 16, 107.
- Wijburg, F.A.; Sedel, F.; Pineda, M.; Hendriksz, C.J.; Fahey, M.; Walterfang, M.; Patterson, M.C.; Wraith, J.E.; Kolb, S.A. Development of a Suspicion Index to Aid Diagnosis of Niemann-Pick Disease Type C. Neurology 2012, 78, 1560–1567.
- Kruth, H.S.; Cupp, J.E.; Khan, M.A. Method for Detection and Isolation of Cholesteryl Ester-containing “Foam” Cells Using Flow Cytometry. Cytometry 1987, 8, 146–152.
- Boernig, H.; Geyer, G. Staining of Cholesterol with the Fluorescent Antibiotic “Filipin”. Acta Histochem. 1974, 50, 110–115.
- Vanier, M.T.; Latour, P. Laboratory Diagnosis of Niemann-Pick Disease Type C: The Filipin Staining Test. Methods Cell Biol. 2015, 126, 357–375.
- Probert, F.; Ruiz-Rodado, V.; Te Vruchte, D.; Nicoli, E.R.; Claridge, T.D.W.; Wassif, C.A.; Farhat, N.; Porter, F.D.; Platt, F.M.; Grootveld, M. NMR Analysis Reveals Significant Differences in the Plasma Metabolic Profiles of Niemann Pick C1 Patients, Heterozygous Carriers, and Healthy Controls. Sci. Rep. 2017, 7, 6320.
- Jiang, X.; Sidhu, R.; Porter, F.D.; Yanjanin, N.M.; Speak, A.O.; Te Vruchte, D.T.; Platt, F.M.; Fujiwara, H.; Scherrer, D.E.; Zhang, J.; et al. A Sensitive and Specific LC-MS/MS Method for Rapid Diagnosis of Niemann-Pick C1 Disease from Human Plasma. J. Lipid Res. 2011, 52, 1435–1445.
- Reunert, J.; Fobker, M.; Kannenberg, F.; Du Chesne, I.; Plate, M.; Wellhausen, J.; Rust, S.; Marquardt, T. Rapid Diagnosis of 83 Patients with Niemann Pick Type C Disease and Related Cholesterol Transport Disorders by Cholestantriol Screening. EBioMedicine 2015, 4, 170–175.
- Boenzi, S.; Deodato, F.; Taurisano, R.; Martinelli, D.; Verrigni, D.; Carrozzo, R.; Bertini, E.; Pastore, A.; Dionisi-Vici, C.; Johnson, D.W. A New Simple and Rapid LC-ESI-MS/MS Method for Quantification of Plasma Oxysterols as Dimethylaminobutyrate Esters. Its Successful Use for the Diagnosis of Niemann-Pick Type C Disease. Clin. Chim. Acta 2014, 437, 93–100.
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; Te Vruchte, D.; Al Eisa, N.; Cortina-Borja, M.; Porter, F.D.; et al. A Novel, Highly Sensitive and Specific Biomarker for Niemann-Pick Type C1 Disease. Orphanet J. Rare Dis. 2015, 10, 78.
- Welford, R.W.D.; Garzotti, M.; Lourenço, C.M.; Mengel, E.; Marquardt, T.; Reunert, J.; Amraoui, Y.; Kolb, S.A.; Morand, O.; Groenen, P. Plasma Lysosphingomyelin Demonstrates Great Potential as a Diagnostic Biomarker for Niemann-Pick Disease Type C in a Retrospective Study. PLoS ONE 2014, 9, e114669.
- Jiang, X.; Sidhu, R.; Mydock-McGrane, L.; Hsu, F.F.; Covey, D.F.; Scherrer, D.E.; Earley, B.; Gale, S.E.; Farhat, N.Y.; Porter, F.D.; et al. Development of a Bile Acid-Based Newborn Screen for Niemann-Pick Disease Type C. Sci. Transl. Med. 2016, 8, 337ra63.
- Mazzacuva, F.; Mills, P.B.; Mills, K.; Platt, F.; Maekawa, M.; Porter, F.D.; Clayton, P.T. Identification of New Biomarkers Suitable for an Early Diagnosis of Niemann-Pick C1. Mol. Genet. Metab. 2016, 117, S78.
- Patterson, M.C.; Clayton, P.; Gissen, P.; Anheim, M.; Bauer, P.; Bonnot, O.; Dardis, A.; Dionisi-Vici, C.; Klünemann, H.H.; Latour, P.; et al. Recommendations for the Detection and Diagnosis of Niemann-Pick Disease Type C: An Update. Neurol. Clin. Pract. 2017, 7, 499–511.
- Maekawa, M.; Jinnoh, I.; Matsumoto, Y.; Narita, A.; Mashima, R.; Takahashi, H.; Iwahori, A.; Saigusa, D.; Fujii, K.; Abe, A.; et al. Structural Determination of Lysosphingomyelin-509 and Discovery of Novel Class Lipids from Patients with Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20, 5018.
- Sidhu, R.; Mondjinou, Y.; Qian, M.; Song, H.; Kumar, A.B.; Hong, X.; Hsu, F.-F.; Dietzen, D.J.; Yanjanin, N.M.; Porter, F.D.; et al. N-Acyl-O-Phosphocholineserines: Structures of a Novel Class of Lipids That Are Biomarkers for Niemann-Pick C1 Disease. J. Lipid Res. 2019, 60, 1410–1424.
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the Diagnosis and Management of Niemann-Pick Disease Type C: An Update. Mol. Genet. Metab. 2012, 106, 330–344.
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Consensus Clinical Management Guidelines for Niemann-Pick Disease Type C. Orphanet J. Rare Dis. 2018, 13, 50.
- Shammas, H.; Kuech, E.M.; Rizk, S.; Das, A.M.; Naim, H.Y. Different Niemann-Pick C1 Genotypes Generate Protein Phenotypes That Vary in Their Intracellular Processing, Trafficking and Localization. Sci. Rep. 2019, 9, 5292.
- Vanier, M.T. Niemann-Pick Disease Type C. Orphanet J. Rare Dis. 2010, 5, 16.
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 Disease: Correlations between NPC1 Mutations, Levels of NPC1 Protein, and Phenotypes Emphasize the Functional Significance of the Putative Sterol-Sensing Domain and of the Cysteine-Rich Luminal Loop. Am. J. Hum. Genet. 2001, 68, 1373–1385.
- Ribeiro, I.; Marcão, A.; Amaral, O.; Sá Miranda, M.C.; Vanier, M.T.; Millat, G. Niemann-Pick Type C Disease: NPC1 Mutations Associated with Severe and Mild Cellular Cholesterol Trafficking Alterations. Hum. Genet. 2001, 109, 24–32.
- Guatibonza Moreno, P.; Pardo, L.M.; Pereira, C.; Schroeder, S.; Vagiri, D.; Almeida, L.S.; Juaristi, C.; Hosny, H.; Loh, C.C.Y.; Leubauer, A.; et al. At a Glance: The Largest Niemann-Pick Type C1 Cohort with 602 Patients Diagnosed over 15 Years. Eur. J. Hum. Genet. 2023, 31, 1108–1116.
- Park, W.D.; O’Brien, J.F.; Lundquist, P.A.; Kraft, D.L.; Vockley, C.W.; Karnes, P.S.; Patterson, M.C.; Snow, K. Identification of 58 Novel Mutations in Niemann-Pick Disease Type C: Correlation with Biochemical Phenotype and Importance of PTC1-like Domains in NPC1. Hum. Mutat. 2003, 22, 313–325.
- Gieselmann, V. Cellular Pathophysiology of Lysosomal Storage Diseases. In Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 190353903X.
- Polten, A.; Fluharty, A.L.; Fluharty, C.B.; Kappler, J.; von Figura, K.; Gieselmann, V. Molecular Basis of Different Forms of Metachromatic Leukodystrophy. N. Engl. J. Med. 1991, 324, 18–22.
- McInnes, B.; Potier, M.; Wakamatsu, N.; Melancon, S.B.; Klavins, M.H.; Tsuji, S.; Mahuran, D.J. An Unusual Splicing Mutation in the HEXB Gene Is Associated with Dramatically Different Phenotypes in Patients from Different Racial Backgrounds. J. Clin. Investig. 1992, 90, 306–314.
- Paron, F.; Dardis, A.; Buratti, E. Pre-MRNA Splicing Defects and RNA Binding Protein Involvement in Niemann Pick Type C Disease. J. Biotechnol. 2020, 318, 20–30.
- Millat, G.; Chikh, K.; Naureckiene, S.; Sleat, D.E.; Fensom, A.H.; Higaki, K.; Elleder, M.; Lobel, P.; Vanier, M.T. Niemann-Pick Disease Type C: Spectrum of HE1 Mutations and Genotype/Phenotype Correlations in the NPC2 Group. Am. J. Hum. Genet. 2001, 69, 1013–1021.
- Tarugi, P.; Ballarini, G.; Bembi, B.; Battisti, C.; Palmeri, S.; Panzani, F.; Di Leo, E.; Martini, C.; Federico, A.; Calandra, S. Niemann-Pick Type C Disease: Mutations of NPC1 Gene and Evidence of Abnormal Expression of Some Mutant Alleles in Fibroblasts. J. Lipid Res. 2002, 43, 1908–1919.
- Macías-Vidal, J.; Gort, L.; Lluch, M.; Pineda, M.; Coll, M.J. Nonsense-Mediated MRNA Decay Process in Nine Alleles of Niemann-Pick Type C Patients from Spain. Mol. Genet. Metab. 2009, 97, 60–64.
- Yamamoto, T.; Nanba, E.; Ninomiya, H.; Higaki, K.; Taniguchi, M.; Zhang, H.; Akaboshi, S.; Watanabe, Y.; Takeshima, T.; Inui, K.; et al. NPC1 Gene Mutations in Japanese Patients with Niemann-Pick Disease Type C. Hum. Genet. 1999, 105, 10–16.
- Encarnação, M.; Coutinho, M.F.; Cho, S.M.; Cardoso, M.T.; Ribeiro, I.; Chaves, P.; Santos, J.I.; Quelhas, D.; Lacerda, L.; Leão Teles, E.; et al. NPC1 Silent Variant Induces Skipping of Exon 11 (p.V562V) and Unfolded Protein Response Was Found in a Specific Niemann-Pick Type C Patient. Mol. Genet. Genom. Med. 2020, 8, e1451.
- Fernandez-Valero, E.M.; Ballart, A.; Iturriaga, C.; Lluch, M.; Macias, J.; Vanier, M.T.; Pineda, M.; Coll, M.J. Identification of 25 New Mutations in 40 Unrelated Spanish Niemann-Pick Type C Patients: Genotype-Phenotype Correlations. Clin. Genet. 2005, 68, 245–254.
- Zhang, X.H.F.; Chasin, L.A. Computational Definition of Sequence Motifs Governing Constitutive Exon Splicing. Genes Dev. 2004, 18, 1241–1250.
- Goren, A.; Ram, O.; Amit, M.; Keren, H.; Lev-Maor, G.; Vig, I.; Pupko, T.; Ast, G. Comparative Analysis Identifies Exonic Splicing Regulatory Sequences-The Complex Definition of Enhancers and Silencers. Mol. Cell 2006, 22, 769–781.
- Nadjar, Y.; Hütter-Moncada, A.L.; Latour, P.; Ayrignac, X.; Kaphan, E.; Tranchant, C.; Cintas, P.; Degardin, A.; Goizet, C.; Laurencin, C.; et al. Adult Niemann-Pick Disease Type C in France: Clinical Phenotypes and Long-Term Miglustat Treatment Effect. Orphanet J. Rare Dis. 2018, 13, 175.
- Anheim, M.; Lagha-Boukbiza, O.; Fleury-Lesaunier, M.C.; Valenti-Hirsch, M.P.; Hirsch, E.; Gervais-Bernard, H.; Broussolle, E.; Thobois, S.; Vanier, M.T.; Latour, P.; et al. Heterogeneity and Frequency of Movement Disorders in Juvenile and Adult-Onset Niemann-Pick C Disease. J. Neurol. 2014, 261, 174–179.
- Ohno, K.; Suzuki, K. Multiple Abnormal Beta-Hexosaminidase Alpha Chain MRNAs in a Compound-Heterozygous Ashkenazi Jewish Patient with Tay-Sachs Disease. J. Biol. Chem. 1988, 263, 18563–18567.
- Caminsky, N.; Mucaki, E.J.; Rogan, P.K. Interpretation of MRNA Splicing Mutations in Genetic Disease: Review of the Literature and Guidelines for Information-Theoretical Analysis. F1000Research 2014, 3, 282.
- Anna, A.; Monika, G. Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 2018, 59, 253–268.
- Sarkar, A.; Panati, K.; Narala, V.R. Code inside the Codon: The Role of Synonymous Mutations in Regulating Splicing Machinery and Its Impact on Disease. Mutat. Res. Rev. Mutat. Res. 2022, 790, 108444.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
596
Revisions:
2 times
(View History)
Update Date:
25 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No